 |  | Theo Dingermann und Christina Hohmann-Jeddi |
 |
13.12.2020 08:00 Uhr |

Weltweit warten und hoffen die Menschen auf eine Eindämmung der Coronavirus-Pandemie. Die neuen Impfstoffe sollen dazu beitragen. / Foto: Adobe Stock/isayurtsever
In der modernen Geschichte wurden noch nie Impfstoffe als so wesentlich für die Wiederherstellung der Normalität in der Gesellschaft angesehen wie die Impfstoffe zum Schutz vor Covid-19. Die Epidemie hat sich schnell zur Pandemie ausgeweitet und das neue Virus SARS-CoV-2 hat keinen Aspekt des Lebens verschont.
Entsprechend hoch waren die Erwartungen an die Pharmaindustrie. Diese hat die Herausforderung angenommen und mit »Warp Speed« oder »Lightspeed« Impfstoffe entwickelt. Offenbar mit Erfolg, wie die bisherigen Wirksamkeitsdaten aus den ersten Phase-III-Studien ganz verschiedener Impfstoffkonzepte zeigen. Innerhalb von zwei Wochen meldeten drei Unternehmen ihre Daten: Die mRNA-Vakzine des Konsortiums Biontech/Pfizer schützt zu 95 Prozent, die mRNA-Vakzine des US-Unternehmens Moderna zu 94,5 Prozent und der Vektorimpfstoff von Astra-Zeneca in Kooperation mit der Universität Oxford zu 62 beziehungsweise 90 Prozent vor einer Covid-19-Erkrankung. Zwei Vakzinen stehen unmittelbar vor der EU-Zulassung, eine hat am 2. Dezember bereits eine Notfallzulassung in Großbritannien erhalten – und das nicht einmal ein Jahr, nachdem das Genom des Erregers veröffentlicht wurde (Kasten).
Foto: Biontech
Vier Impfstoffkandidaten gegen Covid-19 befinden sich bereits im Zulassungsverfahren der Europäischen Arzneimittelagentur EMA. Diese prüft die Vakzinen in einem beschleunigten Verfahren, das als »Rolling Review« bezeichnet wird. Bei diesem werden die Studiendaten nicht zum Abschluss der Studien gesammelt eingereicht, sondern es werden kontinuierlich Zwischenergebnisse geliefert, sodass diese schon parallel zu laufenden Studien geprüft werden können. Klinische Phasen werden nicht übersprungen und es gibt auch sonst keine Änderungen vom regulären Prüfablauf. Auf der Website der EMA heißt es: »Die EMA wird ihre Abschätzung entsprechend der üblichen Standards zu Qualität, Sicherheit und Wirksamkeit vornehmen.«
Beantragt haben die Unternehmen Moderna und Biontech/Pfizer eine bedingte Zulassung (Conditional marketing authorisation). Diese wird für Arzneimittel erteilt, die einen »ungedeckten medizinischen Bedarf« erfüllen, auch wenn die Daten noch nicht vollständig vorliegen. Dies gilt etwa für Wirkstoffe gegen stark einschränkende oder lebensbedrohende Erkrankungen ohne Therapieoption sowie für Arzneimittel, die in Notfallsituationen eingesetzt werden sollen. Die bedingte Zulassung gilt nur für ein Jahr, kann aber jährlich erneuert werden oder in eine Vollzulassung übergehen, informiert das PEI auf Anfrage der PZ.
Eine bedingte Zulassung ist an Auflagen geknüpft. Sie kann erteilt werden, wenn die Nutzen-Risiko-Bilanz positiv ausfällt, also der Nutzen für die öffentliche Gesundheit durch die sofortige Verfügbarkeit des Arzneimittels oder Impfstoffs die Risiken überwiegt, die aufgrund der vorgesehenen Nachreichung weiterer Daten bestehen. Der Antragsteller muss umfassende Daten zu einem späteren Zeitpunkt vorlegen.
Damit unterscheidet sich die bedingte Zulassung von einer Emergency Use Authorization (EUA), eine Notfallgenehmigung, die in den USA das Inverkehrbringen eines nicht zugelassenen Arzneimittels ermöglicht. Eine EUA erlaubt also kein dauerhaftes Inverkehrbringen.
Zulassungen gelten generell nur für die Personengruppen, die in den klinischen Prüfungen vertreten waren, im Fall der SARS-CoV-2-Impfstoffe also nicht für Kinder und Schwangere. Wie Moderna gegenüber der PZ mitteilte, werden entsprechende Studien zu diesen Personengruppen geplant. Bei Schwangeren müssten noch zusätzliche Toxizitätstest bei Tieren abgewartet werden.
Die mRNA-Vakzine von Biontech und Pfizer (Foto) erhielt am 2. Dezember als weltweit erste eine Notfallzulassung von der britischen Arzneimittelbehörde MHRA. Sie ist für Personen ab 16 Jahren zugelassen, denn Jugendliche waren in den Studien vertreten. Auch hier liegen noch keine Daten zu Kindern und Schwangeren vor.
Diese bisher unvorstellbare Geschwindigkeit, mit der diese Impfstoffe entwickelt wurden, ist auf der einen Seite sehr zu begrüßen. Vielen macht sie aber auch Angst, weil einige Vakzinen auf ganz neuen Impfprinzipien basieren. Ganz wichtig ist jetzt Aufklärung. Wie war so eine rasche Entwicklung möglich? Gab es Abstriche bei der Sicherheit?
52 Impfstoffprojekte sind laut Angaben der Weltgesundheitsorganisation (WHO) derzeit in der klinischen Entwicklung, davon 13 in Phase III. Von diesen werden voraussichtlich mehrere in den nächsten Wochen eine Zulassung erhalten. Damit wird sich eine bislang ungekannte Situation einstellen: Erstmals werden Impfstoffe parallel verfügbar sein, die nach ganz unterschiedlichen Prinzipien funktionieren, weil ihre Entwicklungskonzepte extrem unterschiedlich sind. Weit in ihrer Entwicklung sind mRNA-Impfstoffe, Ganzvirus-, Vektor- und Protein-Impfstoffe.
Mit Ausnahme der Ganzvirus-Impfstoffe ist allen Corona-Impfstoffen gemein, dass das Antigen, gegen das die Immunantwort induziert wird, das Spike-Protein (S-Protein) von SARS-CoV-2 ist. Das Coronavirus nutzt dieses Oberflächenprotein, um in Wirtszellen einzudringen.
Nur der Ganzvirus-Impfstofftyp, der zunächst in Deutschland nicht zur Verfügung stehen wird, enthält als Antigen inaktivierte Viren. Diese Impfstoffe werden vor allem in der Volksrepublik China, beispielsweise vom Wuhan Institute of Biological Products, entwickelt beziehungsweise sind dort bereits verfügbar.
Ganz vorne im Rennen liegen die mRNA-Impfstoffe. Das US-Unternehmen Moderna und das Konsortium Biontech/Pfizer haben für ihre Covid-19-Impfstoffe bereits am 30. November einen Antrag auf bedingte Zulassung in der EU gestellt. Die Vakzinen enthalten einzelsträngige, in Lipid-Nanopartikel verpackte Boten-RNA (mRNA), die für das S-Protein kodiert. Im Körper des Geimpften müssen die Moleküle in Zellen gelangen und in das eigentliche Antigen übersetzt werden. Die mRNA-Impfstoffe stellen ein ganz neues Prinzip dar und sind daher die innovativste Form unter den Impfstoffkandidaten. »Innovativ« bedeutet im Gesundheitsbereich aber immer auch »wenig erprobt«. Und hier liegt wohl ein Schwachpunkt dieser Impfstoffklasse, die ansonsten gute Eigenschaften hat.
Die Vorteile: mRNA-Impfstoffe lassen sich schnell produzieren und auch schnell modifizieren, falls dies durch Mutationen des Virus erforderlich werden sollte. Die Produkte enthalten kein toxisches oder tierisches Material aus dem Herstellungsprozess. Zudem benötigen sie keine Vektorviren, die störende Reaktionen induzieren könnten, um die genetische Information zu transportieren. mRNA-Impfstoffe regen das Immunsystem des Impflings zu einer starken Bildung neutralisierender Antikörper an und induzieren zudem die Bildung spezifischer zytotoxischer CD8+-Zellen. In bisherigen Auswertungen der Phase-III-Studien zeigten die Kandidaten von Moderna und Biontech/Pfizer eine überraschend hohe Wirksamkeit.
Da fremde mRNA selbst das Immunsystem stimuliert, kommen die Impfstoffe in der Regel ohne Adjuvans aus. Bei den Kandidaten, die weit in der Entwicklung sind, ist aber ein Prime-Boost-Schema, also die Gabe von zwei Dosen innerhalb von drei beziehungsweise vier Wochen, erforderlich.

Der Stoff, aus dem die Träume sind: mRNA hat die Impfstoffforschung beflügelt. / Foto: Adobe Stock/nobeastsofierce
Die Nachteile: Das Impfstoffprinzip ist wenig erprobt. Langzeitdaten gibt es nicht. In Studien wurden aber die akuten Nebenwirkungen quantifiziert, die allerdings vorübergehend und meist mild bis moderat sind. So kam es bei mRNA-1273 von Moderna vor allem zu Reaktionen wie Schmerzen und Rötung an der Einstichstelle, Fatigue, Muskel-, Gelenk- und Kopfschmerzen. Die Immunisierung mit BNT162b2 von Biontech/Pfizer wurde in einer Phase-I-Studie in allen Populationen gut vertragen; weniger als 20 Prozent der Probanden hatten leichtes bis moderates Fieber. Insgesamt waren die Nebenwirkungen nach der ersten Dosis BNT162b2 bei Probanden der Altersgruppe 65 bis 85 Jahre vergleichbar mit den Nebenwirkungen in der Placebogruppe. Nach der zweiten Impfung à 30 µg mit BNT162b2 trat bei 17 Prozent der Probanden zwischen 18 und 55 Jahren und bei 8 Prozent der Probanden zwischen 65 und 85 Jahren Fieber auf (≥ 38 bis 38,9 °C). Dagegen waren Kälteschauer (Chills) und Fatigue mit bis zu 80 Prozent häufig. Lokale Reaktionen wie Schmerzen an der Injektionsstelle zeigten fast alle Probanden.
Ein Problem ist, dass der Impfstoff BNT162b2 bei sehr niedrigen Temperaturen von -60 bis -80 °C gelagert und transportiert werden muss und eine spezielle Logistik benötigt. Dies scheint aber nicht an der Molekülart mRNA zu liegen, denn die Produkte von Moderna und dem Tübinger Unternehmen Curevac, das ebenfalls an RNA-Impfstoffen arbeitet, stellen diese hohen Ansprüche nicht.
Befürchtungen, dass die mRNA in der Zelle zu DNA revers transkribiert wird und dann in das Wirtsgenom integriert werden könnte, sind theoretischer Natur. Bei mRNA-Impfstoffen handelt es sich nicht um Gentherapeutika. Eine Mutagenität wird, wie bei allen neuen Wirkstoffen, präklinisch untersucht.
Statt die genetische Information für das S-Protein »nackt« verpackt in Lipid-Nanopartikeln zu verimpfen, ist sie bei Vektorimpfstoffen in das Genom von abgeschwächten DNA-Viren integriert, die als Transportvehikel in die menschlichen Zellen fungieren. Dort wird die Information für das Antigen abgelesen, das S-Protein gebildet und dem Immunsystem präsentiert. Momentan stehen folgende virale Vektoren, die alle replikationsinkompetent sind, im Fokus: humane Adenoviren (AdV/zum Beispiel Janssen), ein Schimpansen-Adenovirus (ChAdOx/Universität Oxford mit Astra-Zeneca) und ein modifiziertes Vaccinia-Ankara-Virus (MVA/LMU München mit dem Deutschen Zentrum für Infektionsforschung).
Die Vorteile: Vektorimpfstoffe regen wie die mRNA-Vakzinen neben einer Antikörper-Antwort auch eine potente CD8+-T-Zellantwort an. Sie benötigen ebenfalls kein Adjuvans, in der Regel aber ein Prime-Boost-Schema. Gegenüber den mRNA-Impfstoffen haben sie den Vorteil, dass dieser Impfstofftypus nicht mehr ganz neu ist: Einige Vektorvakzinen gegen verschiedene Erreger sind bereits zugelassen.
Die Nachteile: Es ist gut belegt, dass durch eine bereits bestehende Immunität gegen das Vektorvirus eine erneute Immunisierung neutralisiert werden kann. Das liegt daran, dass nicht nur eine Immunantwort gegen das S-Protein von SARS-CoV-2 induziert wird. Es werden darüber hinaus auch Antikörper und T-Zellen gegen die Proteine produziert, die das Vektorgenom kodiert. So besteht die Gefahr, dass es bei einer weiteren Anwendung zu einem vorzeitigen Abbau des Impfstoffs oder zu überschießenden Immunreaktionen dagegen kommen kann.
| Name | Hersteller | Impfstofftyp | Impfschema | Lagerung | Start der Prüfung bei der EMA |
|---|---|---|---|---|---|
| BNT162b2 | Biontech/Pfizer | mRNA | Tag 0, Tag 21 | -70 bis -80 °C | 6. Oktober |
| mRNA-1273 | Moderna | mRNA | Tag 0, Tag 28 | -20 °C | 16. November |
| AZD1222 | Astra-Zeneca/Universität Oxford | Vektor | Tag 0, Tag 28 | 2 bis 8 °C | 1. Oktober |
| Ad26.COV2.S | Janssen | Vektor | Tag 0 | 2 bis 8 °C | 1. Dezember |
Das bedeutet, dass man im Extremfall auf eine Vorimmunität testen müsste, um diese Impfstoffe effektiv und sicher einsetzen zu können. Und wahrscheinlich ist es auch so, dass jeder einzelne Vektortyp nur einmal pro Impfling angewendet werden kann. Wer also bereits einen Ad26-Vektor-Impfstoff erhalten hat, könnte in Zukunft nicht mit dem gleichen Vektor (mit anderer »Beladung«) gegen eine andere Erkrankung geimpft werden.
Der russische vektorbasierte Sputnik-Impfstoff sorgte weltweit für Aufsehen, als er am 11. August 2020 als erster Coronavirus-Impfstoff überhaupt zugelassen wurde – ganz ohne Phase-III-Studie. / Foto: kandinskiy_/Shotshop.com
Dem tragen verschiedene Hersteller dieses Impfstofftyps Rechnung. So werden zum Beispiel für eine Impfung mit dem russischen Sputnik-V-Impfstoff zwei unterschiedliche Impfstofftypen für die Prime- und die Boost-Impfung eingesetzt, nämlich Ad26 gefolgt von Ad5. Das Unternehmen Janssen, die Pharmasparte von Johnson & Johnson, benötigt für seinen Impfstoffkandidaten Ad26.COV2.S nur eine Dosis und umgeht damit das Problem. Dagegen hat sich das Konsortium der Universität Oxford und Astra-Zeneca entschlossen, ihren Impfstoffkandidaten AZD1222 (mit einem Schimpansen-Adenovirus) sowohl bei der Prime- als auch bei der Boost-Impfung einzusetzen.
Die Wirksamkeit von Sputnik V liegt laut einer Zwischenauswertung bei mehr als 95 Prozent. Für AZD1222 wurden je nach Impfregime eine Wirksamkeit von 62 beziehungsweise 90 Prozent ermittelt. Auf den ersten Blick erstaunlich ist, dass die geringere Wirksamkeit von 62 Prozent bei einem Impfregime gemessen wurde, bei dem bei beiden Impfungen (Prime und Boost) die volle Dosis von 5 x 1010 Viruspartikeln appliziert wurde. Ein deutlich besseres Ergebnis erhielt man, wenn bei der Prime-Impfung die halbe Dosis verabreicht und dann bei der Boosterung die volle Dosis appliziert wurde. Hier bestätigt sich empirisch die Theorie, dass ein vorhandener Immunschutz vor dem Vektorvirus eine weitere Impfung mit dem gleichen Vektor ineffizient machen könnte, da das Immunsystem den Vektor attackiert. Die genauen Gründe für den Befund werden noch untersucht.
Als denkbarer Nachteil gilt auch eine zufällige Integration der Vektor-DNA in das Genom des Wirts. Dies könnte eine verstärkte Tumorbildung infolge einer Aktivierung von Onkogenen oder Deaktivierung von Tumorsuppressor-Genen induzieren oder es könnten Autoimmunkrankheiten entstehen. Diese These sieht der Präsident des Paul-Ehrlich-Instituts, Professor Dr. Klaus Cichutek, allerdings als entkräftet an. Bei einem Pressebriefing des »Science Media Center Germany« am 27. April sagte er: »Wir haben bei den DNA-Impfstoffen lange Jahrzehnte damit verbracht, einem theoretischen Risiko nachzugehen, das sich dann am Tier und in klinischen Prüfungen eigentlich nie bewahrheitet hat.«
Ein deutlich konservativerer Ansatz sind Protein-Impfstoffe. Sie enthalten gentechnisch hergestelltes S-Protein, analog zum Hepatitis-B-Impfstoff, der Hepatitis-B-Oberflächenantigen enthält, oder zum HPV-Impfstoff, der als Antigen L1-Proteine verschiedener Typen des humanen Papillomavirus beinhaltet. Ein SARS-CoV-2-Impfstoff nach diesem Prinzip befindet sich bereits in Phase III der klinischen Entwicklung, nämlich der Kandidat des US-Unternehmens Novavax, der mit Matrix-M1 adjuvantiert ist. In Phase I/II befindet sich die Protein-Vakzine des Konsortiums Sanofi/Glaxo-Smith-Kline (GSK). Eine Phase-III-Studie soll noch in diesem Jahr beginnen.
Aktuell startet in Deutschland auch eine klinische Phase-I-Studie mit einem adjuvantierten Peptid-Impfstoff, der an der Universität Tübingen entwickelt wurde. Antigene sind bei ihm Bruchstücke (Peptide) von mehreren SARS-CoV-2-Proteinen, darunter das S- und das Nukleokapsid-Protein.
Die Vorteile: Das Prinzip der Protein-Impfstoffe ist bekannt. Dieser Impfstofftyp induziert sehr gut die Bildung neutralisierender Antikörper.
Die Nachteile: Die T-Zell-Antwort ist eher schwach. Daher muss diesem Impfstofftyp mit Sicherheit ein Adjuvans zugesetzt werden. Gegen diese gibt es seit der H1N1-Influenza-Pandemie (»Schweinegrippe«) im Jahr 2009/2010 gewisse Bedenken, da der mit AS03 adjuvantierte Pandemie-Impfstoff Pandemrix® zu einer relevanten Erhöhung des Risikos für die Autoimmunerkrankung Narkolepsie führte. Der genaue Pathomechanismus und ob das Adjuvanz beteiligt war, ist nicht vollständig aufgeklärt. Die Covid-19-Vakzine von Sanofi soll den Wirkverstärker AS03 von GSK enthalten, bestätigte GSK gegenüber der Pharmazeutischen Zeitung.
Bisher dauerte die Entwicklung von Impfstoffen etwa 15 Jahre, bei Covid-19 voraussichtlich nur knapp ein Jahr (Grafik). Warum die Entwicklung für diesen Erreger so schnell ging, erklärte Professor Dr. Stephan Becker vom Institut für Virologie an der Philipps Universität Marburg bei einer Veranstaltung von »House of Pharma & Healthcare« am 26. November. Ein Grund sei die Ebola-Epidemie von 2014 bis 2016, in deren Verlauf 11.000 Menschen starben. Damals wurden rasch Impfstoffe gegen das Ebolavirus entwickelt, von denen aber bald klar war, dass sie für das Eindämmen des Ausbruchs zu spät kommen würden, sagte Becker. Inzwischen sind zwei Vektorimpfstoffe gegen das Ebolavirus zugelassen: Ervebo® von MSD (November 2019) und die Kombination der zwei Vektorimpfstoffe Zabdeno® und Mvabea® von Janssen-Cilag (Mai 2020).
Die Ebola-Epidemie sei ein Weckruf gewesen, der dazu führte, dass die Impfstoffforschung zu Erregern mit pandemischen Potenzial verstärkt wurde. Hierzu wurde 2017 die Coalition for Epidemic Preparedness Innovations (CEPI) gegründet, eine weltweite Allianz von Regierungen, der WHO, der Pharmaindustrie und privaten Geldgebern, um auf zukünftige Pandemien schneller reagieren zu können. Diese wurde mit einer Milliarde US-Dollar ausgestattet, berichtete Becker. So war es möglich, schon im Vorfeld Impfstoffkandidaten gegen hochpathogene Viren wie das Lassavirus sowie die zwei Coronaviren MERS-CoV (das 2012 erstmals auftrat) und SARS-CoV-1 (das 2002 und 2004 einen Ausbruch verursachte) zu entwickeln.
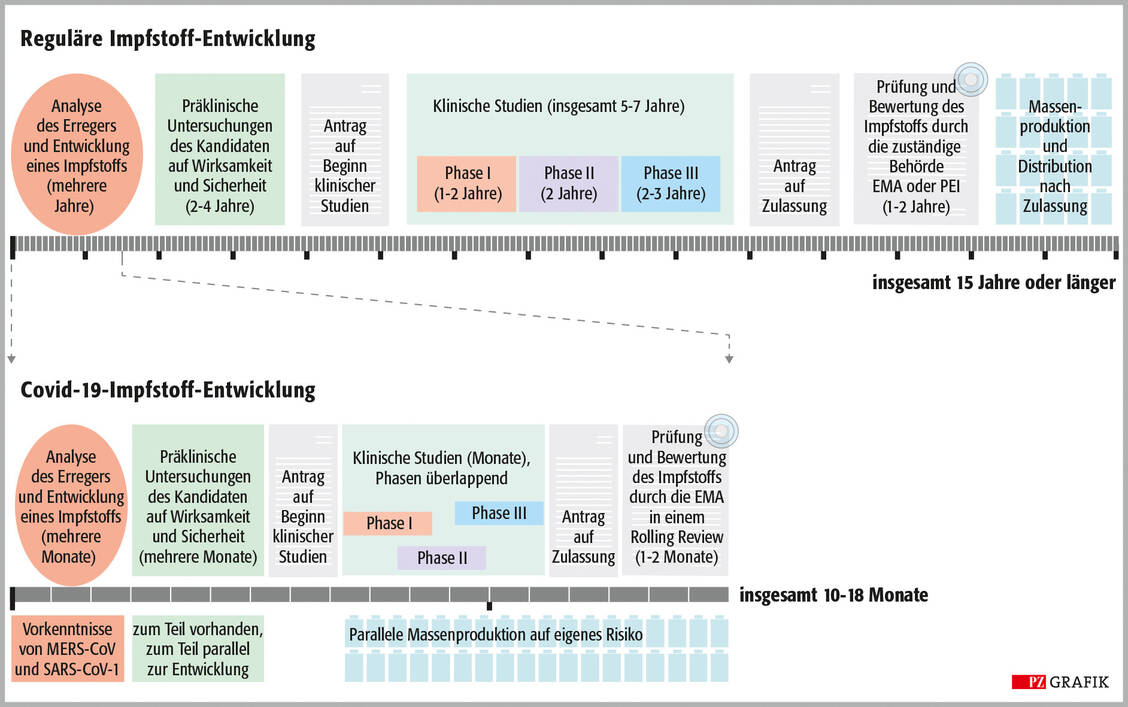
Grafik: Zeitlicher Ablauf der regulären Impfstoff-Entwicklung und der Covid-19-Impfstoff-Entwicklung im Vergleich. Der reguläre Prozess (oben) kann bis zu 15 Jahre dauern; mit Erteilung der Zulassung beginnen die Massenproduktion und das Inverkehrbringen des Produkts. Bei den Impfstoffen gegen Covid-19 konnte dieser Prozess auf etliche Monate abgekürzt werden. Die EMA-Zulassung der ersten Präparate wird demnächst erwartet. Modifiziert nach »Nature« 2020, DOI: 10.1038/s41586-020-2798-3 / Foto: PZ/Stephan Spitzer
Seit Auftreten der beiden nah verwandten Coronaviren wird an Impfstoffen gegen die Pathogene gearbeitet. Auch Beckers Arbeitsgruppe hat zusammen mit der Ludwig-Maximilians-Universität München, dem Universitätsklinikum Hamburg-Eppendorf (UKE) und dem Deutschen Zentrum für Infektionsforschung (DZIF) einen Vektorimpfstoff auf MVA-Basis gegen MERS-CoV entwickelt. Diese Vakzine wurde bei Mäusen, Kamelen und anschließend auch in klinischen Studien auf Immunogenität und Sicherheit geprüft. »Anfang des nächsten Jahres soll eine klinische Wirksamkeitsstudie beginnen«, sagte der Virologe.
Diese Vorarbeit habe viel Zeit bei der Entwicklung der SARS-CoV-2-Impfstoffe gespart. Man hatte nicht nur bereits Impfstoffplattformen etabliert, sondern kannte auch schon die Zielstruktur: das S-Protein der Coronaviren, berichtete Becker. Außerdem konnte man in den präklinischen Untersuchungen auf etablierte Tiermodelle zurückgreifen. Das habe die präklinische Phase, die sonst mehrere Jahre dauert, auf einige Monate abgekürzt (Grafik). Die klinischen Studien konnten somit früh beginnen und liefen zum Teil parallel. So wurden mitunter Phase-I- und -II-Studien zusammengefasst und Phase-III-Studien schon geplant, während die Phase-I/II-Studien noch liefen. »Sobald Sicherheitsprüfungen von unabhängigen Data Safety Monitoring Boards vorlagen, ging es in die nächste Runde«, berichtete der Virologe.

Die internationale Kooperation – und Konkurrenz – von Wissenschaftlern und Pharmaunternehmen hat die Entwicklung enorm vorangetrieben. / Foto: Adobe Stock/Mongkolchon
Das Besondere bei den Covid-19-Impfstoffen: Die Unternehmen starteten eine Massenproduktion schon während der klinischen Studien auf eigenes Risiko. Normalerweise warten sie hierfür ab, bis die Zulassung oder zumindest ausreichend Daten aus Phase III vorliegen.
Zusätzlich wurde noch der regulatorische Zulassungsprozess beschleunigt, der bei anderen Impfstoffen ein bis zwei Jahre dauern kann. Die Covid-19-Impfstoffe werden von der Europäischen Arzneimittelagentur (EMA) in einem sogenannten Rolling-Review-Verfahren geprüft, bei dem die Studiendaten nicht am Ende gesammelt, sondern – sobald sie vorliegen – kontinuierlich eingereicht und entsprechend geprüft werden. Außerdem werden diese Impfstoffe in der Behörde mit höchster Priorität bearbeitet, weshalb die Prüfung wohl nur einen bis zwei Monate dauern wird. »Zusammengenommen führt dies zu einer Entwicklungszeit von etwa einem Jahr«, sagte Becker.
Eine so schnelle Entwicklung kann auch Angst machen und Fragen zur Sicherheit aufwerfen. Entsprechend steht ein Teil der Bevölkerung den neuen Impfstoffen skeptisch gegenüber. Das Ausmaß der Impfskepsis in Deutschland erfasst das Covid-19 Snapshot Monitoring (COSMO), ein Projekt der Universität Erfurt in Kooperation mit dem Robert-Koch-Institut, der Bundeszentrale für gesundheitliche Aufklärung und weiteren Partnern. Seit April erhoben die Wissenschaftler Daten zu verschiedenen Themen, unter anderem der Impfbereitschaft für eine hypothetische Impfung gegen Covid-19. Die Impfbereitschaft sank von 79 Prozent Mitte April auf 54 Prozent Mitte November. Sie hängt der Erhebung zufolge stark vom Vertrauen in die Impfstoffe und vom Ergebnis der Nutzen-Risiko-Bewertung ab. Die Befragung zeigte auch, dass die Kenntnisse über die Impfstoffe noch recht gering sind.
Wer wird sich impfen lassen? Noch ist das Vertrauen der Menschen in die neuen Impfstoffe nicht allzu groß. / Foto: Adobe Stock/Rido
Dass eine persönliche Risiko-Nutzen-Bewertung für die Impfentscheidung eine große Rolle spielt, zeigt auch eine Untersuchung aus Israel. In dieser befragten Dr. Amiel Dror und seine Kollegen von der Bar-Ilan-Universität in Safed, Israel, 1941 Personen, davon 829 Beschäftigte aus dem Gesundheitswesen, ob sie sich gegen Covid-19 impfen lassen würden. Das Ergebnis: Menschen, die sich selbst als Risikopersonen ansahen, und medizinisches Personal, das mit Covid-19-Patienten arbeitete, waren offener für eine Impfung. Insgesamt zeigte sich beim medizinischen Personal aber eine deutliche Skepsis: So war die Impfbereitschaft bei Ärzten mit 78 Prozent knapp über dem Bevölkerungsdurchschnitt (75 Prozent), bei Krankenschwestern lag sie aber mit 61 Prozent deutlich darunter.
Nach den Gründen gegen eine Impfung gefragt, gaben 76 Prozent eine fragliche Sicherheit der Impfstoffe an und 13 Prozent stellten deren Wirksamkeit infrage. Zudem zweifelten 11 Prozent die Gefährlichkeit von SARS-CoV-2 an. Die Autoren betonen, dass gezielte Aufklärungskampagnen vor allem in den Risikogruppen hilfreich wären, um die Impfbereitschaft zu fördern. Hier sei noch viel Aufklärung zu den Impfstoffen nötig, betonte auch Becker. »Gerade beim medizinischen Personal ist es wichtig, dass es sich impfen lässt.«
Auch Bundesforschungsministerin Anja Karliczek (CDU) sieht in der Bevölkerung insgesamt beim Thema Corona-Impfungen noch viel Aufklärungsbedarf. Etwa 50 Prozent Impfbereitschaft seien gut, »es wäre allerdings schön, wenn die Bereitschaft noch etwas steigen würde«, sagte sie der Deutschen Presseagentur. Nach Angaben der WHO ist eine Durchimpfungsrate von 60 bis 70 Prozent der Bevölkerung nötig, um die Corona-Pandemie einzudämmen. Karliczek bekräftigt aber, dass es keinen Impfzwang geben werde. »Es bleibt dabei: Die Impfung wird freiwillig sein.« Die Impfung habe nicht nur einen Vorteil für jede Person selbst, die Impfung sei auch ein Dienst an der Gemeinschaft.
Theo Dingermann studierte Pharmazie in Erlangen. Nach der Approbation 1976 folgten Promotion und 1987 Habilitation. Von 1991 bis 2013 war er Geschäftsführender Direktor des Instituts für Pharmazeutische Biologie an der Goethe-Universität Frankfurt am Main. Jetzt ist er Seniorprofessor der Universität. Dingermann war von 2000 bis 2004 Präsident der DPhG und arbeitet in zahlreichen Gremien, unter anderem beim BfArM. Die Apotheker kennen ihn als Referenten, Autor und Co-Autor von wissenschaftlichen Fach- und Lehrbüchern. Seit April 2010 ist er externes Mitglied der Chefredaktion der PZ, seit Frühjahr 2019 einer der drei Chefredakteure.
Christina Hohmann-Jeddi studierte Biologie an der Johannes-Gutenberg-Universität in Mainz. Nach Abschluss des Studiums absolvierte sie eine Ausbildung zur Wissenschaftsredakteurin, danach machte sie ein Volontariat bei der Pharmazeutischen Zeitung. Seit 2003 leitet sie das Ressort Medizin.


