 |  | Bettina Wick-Urban |
 |
13.03.2022 08:00 Uhr |
Die häufigste monogenetische Erkrankung vor allem in ehemaligen Malariagebieten im Mittelmeerraum, im Vorderen Orient sowie in Afrika und Asien ist die Thalassämie. Etwa 60.000 Neugeborene kommen jedes Jahr mit einer schweren Form der Erkrankung auf die Welt. In Deutschland ist die Erkrankung mit circa 500 Patienten mit einer schwerwiegenden Form sehr selten. Ursache ist eine Mutation oder Deletion in den Genen für die α- oder β-Untereinheit des Hämoglobin, wodurch kein funktionsfähiges Hämoglobin in den Erythrozyten produziert wird (20, 21).
Bei der α-Thalassämie befindet sich der Defekt auf Chromosom 16, das für die α-Untereinheit kodiert. Der Schweregrad der Erkrankung hängt davon ab, wie viele Allele betroffen sind. Bei der schwersten Form sind alle vier Allele des α-Gens inaktiv; unbehandelt stirbt der Embryo bereits intrauterin. Wenn drei Allele inaktiv sind, liegt die sogenannte HbH-Krankheit vor. Dies ist eine leichtere Form der Thalassämie, bei der die Patienten eine hämolytische Anämie aufweisen sowie Leber und Milz vergrößert sind, viele jedoch klinisch kaum Symptome haben und selten Bluttransfusionen benötigen, da noch Hämoglobin gebildet werden kann. Im Erwachsenenalter können Komplikationen wie kardiale Probleme, Gallensteine, Unterschenkelgeschwüre und Folsäuremangel auftreten.
Die β-Thalassämie ist die häufigere Form. Verantwortlich sind Mutationen des Gens auf dem Chromosom 11, das die β-Untereinheit des Hämoglobins kodiert. Bekannt sind mehr als 4000 verschiedene Mutationen, die meist autosomal-rezessiv vererbt werden.
Bei der Thalassämie minor ist nur ein Genallel betroffen und die Patienten haben meist keine klinischen Symptome außer einer leicht vergrößerten Milz. Diese Form ist nicht behandlungsbedürftig.
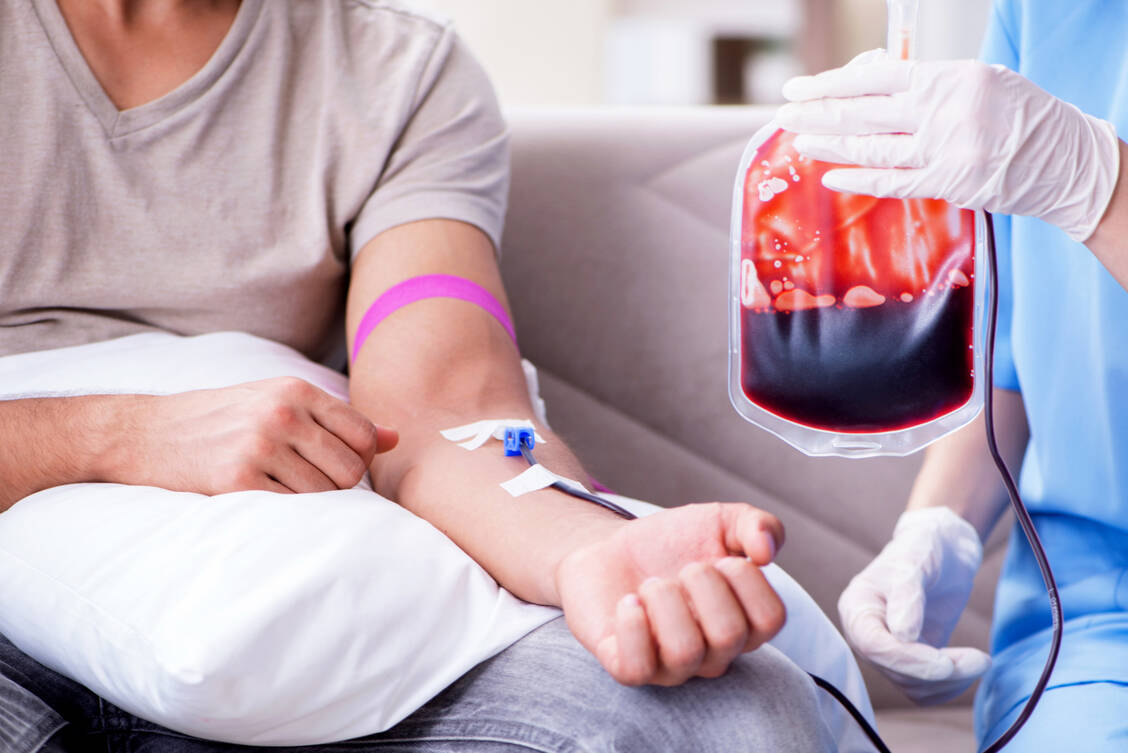
Patienten mit Thalassämien können kein funktionsfähiges Hämoglobin bilden. Je nach Schweregrad der Erkrankung brauchen sie regelmäßige Infusionen von Erythrozytenkonzentraten. / Foto: Shutterstock/Elnur
Bei der Thalassämie major, bei der beide Allele betroffen sind, werden die β-Globinketten nicht synthetisiert und es kann kein normales HbA1 (α2β2) produziert werden. Durch den starken Überschuss an α-Globinen entstehen defekte instabile Erythrozyten, die bereits im Knochenmark zugrunde gehen. Bereits wenige Monate nach der Geburt vergrößern sich Leber und Milz stark. Im weiteren Verlauf treten Wachstumsstörungen, schwere Schäden innerer Organe und Knochenfehlbildungen auf. Durch den Mangel an Erythrozyten und Hämoglobin kann Sauerstoff nicht effektiv transportiert werden: Eine schwere Anämie ist die Folge. Unbehandelt führt die Erkrankung im frühen Kindesalter zum Tod (20, 21).
Standardbehandlung bei β-Thalassämie major ist die Gabe von Erythrozytenkonzentraten sowie eine begleitende Chelattherapie (Deferasirox oder Dexferoxamin), um eine Eisenüberladung zu verhindern, die zu Organschäden, zum Beispiel am Herzen, führen kann. Gibt es einen HLA-identischen Spender, beispielsweise ein Geschwisterkind, ist eine hämatopoetische Stammzelltransplantation die Therapie der Wahl (20).


