 |  | Bettina Wick-Urban |
 |
13.03.2022 08:00 Uhr |
Gentherapeutische Ansätze bei Hämophilie-Patienten verwenden fast ausschließlich in vivo verabreichte rekombinante nicht pathogene replikationsdefekte AAV-Vektoren (Kasten). Diese enthalten ein gesundes Gen für Faktor VIII oder IX sowie eine Hepatozyten-spezifische Promotorsequenz, die die Expression des episomalen Gens in den Leberzellen ermöglicht, da sowohl Faktor VIII als auch Faktor IX physiologisch in der Leber synthetisiert werden. Mehrere Gentherapien werden derzeit klinisch erprobt (9).
Bei Patienten mit Hämophilie A ist die klinische Testung von Valoctocogen roxaparvovec (AAV5-hFVIII-SQ, Valrox®) am weitesten fortgeschritten (Tabelle). Das Transgen ist eine komplementäre einzelsträngige DNA des Faktors VIII, der die B-Domäne fehlt, was die biologische Aktivität nicht vermindert (hFVIII-SQ). Das produzierte Protein hat dieselbe Aminosäuresequenz wie rekombinante Gerinnungsfaktoren, die kein erhöhtes Risiko für Alloantikörper zeigen (Beispiel: ReFacto®).
| Name, Firma | Transgen | Vektor | Klinische Entwicklung |
|---|---|---|---|
| Hämophilie A | |||
| Valoctocogen roxaparvovec (Valrox®), Biomarin | ssFVIII-SQ | AAV5 | Phase III abgeschlossen, Zulassungsantrag eingereicht |
| Giroctocogen fitelparovec, Sangamo/Pfizer | ssFVIII-SQ | AAV2/6 | Phase III |
| SPK-8011, Spark | ssFVIII-SQ | modifizierter AAV3 | Phase II |
| BAY2599023 (DTX201), Bayer/Ultragenyx | ssFVIII-SQ | AAVhu37 | Phase I/II |
| BAX 888, Baxalta/Shire | ssFVIII-SQ | AAV8 | Phase I/II |
| AAV2/8-HLP-FVIII-V3, University College of London | ssFVIII-V3 | AAV2/8 | Phase I |
| CD68-ET3-LV CD34+, Expression Therapeutics | ET3 | Lentivirus | Phase I |
| Hämophilie B | |||
| Etranacogen dezaparvovec, UniQure, CSL Behring | ssFIX-R338L | AAV5 | Phase III |
| Fidanacogen elaparvovec (RAAV-SPARK100-HFIX-PADUA), Pfizer/Spark | ssFIX-R338L | SPK100 | Phase III |
| Verbrinacogen setparvovec, Freeline | FLT180a FIX Padua | AAVS3 | Phase I/II |
In der Phase-III-Studie GENE 8-1 mit 134 Patienten reduzierte eine einmalige Infusion die jährliche Blutungsrate (ABR) um 84 Prozent im Vergleich zur Gabe von Faktor-VIII-Konzentrat und den Bedarf an Faktor VIII um 99 Prozent. Nach zwei Jahren war die produzierte Menge an Faktor VIII geringer, reichte jedoch noch aus für eine hämostatische Wirksamkeit (ABR 0,9). Die Gentherapie wurde gut vertragen; thromboembolische Nebenwirkungen traten nicht auf und es wurden keine Antikörper gegen Faktor VIII gebildet. Erhöhte Leberenzymwerte waren die häufigste Nebenwirkung, jedoch vorübergehend und ohne klinische Symptome. Bei den Zulassungsbehörden in den USA und in Europa (FDA und EMA) sind Zulassungsanträge eingereicht (10).
In einer sehr frühen klinischen Erprobungsphase befindet sich ein gentherapeutischer Ex-vivo-Ansatz. Dabei werden dem Patienten CD34+-Stammzellen entnommen. Mithilfe eines Monozyten-spezifischen Lentivirus wird ex vivo ein hochaktives Faktor-VIII-Gen eingeschleust. Der Patient erhält eine Chemotherapie mit Busulfan und danach die modifizierten Stammzellen als Infusion. Mit Studienergebnissen wird frühestens 2025 gerechnet (11).
Auch für Hämophilie B sind mehrere Gentherapien in der klinischen Erprobung. Am weitesten fortgeschritten ist Etranacogen dezaparvovec (Tabelle). Das verwendete Transgen ist ein physiologisch vorkommendes mutiertes Faktor-IX-Gen. Die Missense-Mutation erhöht die Aktivität des Proteins um das Achtfache im Vergleich zum Wildtyp-Faktor IX.
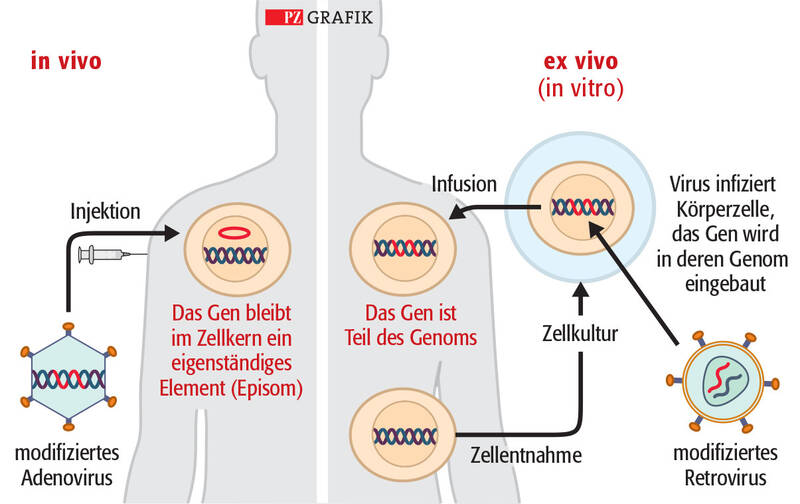
Foto: PZ/Stephan Spitzer
Um genetische Information in die Zielzellen einzuschleusen, werden virale Vektoren verwendet. Diese sogenannte Transduktion kann ex vivo oder in vivo erfolgen. Für eine Gentherapie werden zwei Arten von Viren eingesetzt: Retroviren wie Lentiviren beziehungsweise Adeno- oder Herpesviren.
Um ein Virus als Vektor einzusetzen, muss es zunächst genetisch so modifiziert werden, dass nach Integration des therapeutischen Gens keine Viruspartikel gebildet werden, also das Virus sich nicht repliziert. Zu diesem Zweck werden die entsprechenden viralen Abschnitte des Erbmaterials entfernt.
Bei der Ex-vivo-Gentherapie werden dem Patienten die Zielzellen zunächst entnommen. Mithilfe von Lentiviren, die sich nicht vermehren können, wird das therapeutische Gen stabil in die entnommenen Zellen integriert; das heißt, dass das Gen bei der Zellteilung an die Tochterzellen weitergegeben wird. Anschließend werden die in Zellkultur vermehrten modifizierten Zellen dem Patienten wieder verabreicht. Das Verfahren ist für Zellen geeignet, die sich relativ leicht aus dem Körper gewinnen lassen, zum Beispiel Stammzellen aus dem Knochenmark, und die sich anschließend replizieren.
Bei einer In-vivo-Gentherapie werden Viren, die das therapeutische Gen enthalten, direkt in den Körper des Patienten infundiert oder injiziert. Derzeit werden vor allem Adeno-assoziierte Viren (AAV) als Genfähren verwendet. Die AAV-Vektoren integrieren ihr Erbgut nicht in das Genom der menschlichen Zelle, sondern es verbleibt als sogenanntes Episom im Zellkern und wird während einer Zellteilung daher nicht vermehrt. AAV-Vektoren werden deshalb für das Einbringen von Genen in postmitotisches Gewebe eingesetzt, das sich nicht vermehrt, zum Beispiel Retina, Leber oder Muskel. Die Selektivität der Viren für bestimmte Gewebe wird durch modifizierte Virushüllen und gewebespezifische Promotoren verbessert (8).
In der Phase-III-Studie HOPE-B erhielten 54 Patienten mit schwerer Hämophilie B zunächst sechs Monate ihre Standardtherapie, danach einmal eine Infusion Etranacogen dezaparvovec. Zwölf Monate nach der Infusion betrug die Blutungsrate 1,5; im Vergleich dazu lag die Blutungsrate bei 4,2 während der ersten sechs Monate unter Standardtherapie. Auch 18 Monate nach der Infusion hatten die Patienten eine klinisch relevante Faktor-IX-Aktivität. Die Gentherapie wurde gut vertragen; rund 80 Prozent der Nebenwirkungen waren mild. Faktor-IX-Antikörper traten nicht auf. Die Zulassungsunterlagen sollen 2022 bei den Behörden eingereicht werden (12).
Auch für Fidanacogen elaparvovec liegen ermutigende Ergebnisse einer Phase-II-Studie mit zehn Männern vor. Mithilfe eines Adeno-assoziierten Vektors wurde ein hochaktives Faktor-IX-Transgen infundiert. Die jährliche Blutungsrate sank von 11,1 auf 0,4 nach der Infusion. Die Faktor-IX-Aktivität lag bei 30 Prozent. Während der 18-monatigen Nachbeobachtungszeit traten bei neun der zehn Probanden keine Blutungen auf; acht benötigten keine Gerinnungsfaktoren und die anderen beiden nur in geringem Maß. Die Therapie wurde gut vertragen. Weder Thrombosen oder erhöhte Laborwerte noch gegen Faktor IX gerichtete Antikörper wurden beobachtet (13). Die zulassungsrelevanten Studien zur Langzeitsicherheit und -wirksamkeit dauern noch an.


