 |  | Bettina Wick-Urban |
 |
13.03.2022 08:00 Uhr |

Für etliche monogenetische Erkrankungen, darunter die Hämophilien, gibt es gentherapeutische Ansätze, die Hoffnung auf Heilung wecken. Jedoch sind noch viele Fragen ungeklärt. / Foto: Adobe Stock/RFBSIP
Bei genetischen Erkrankungen liegen Chromosomenstörungen vor, die in der Regel von den Eltern an das Kind weitergegeben werden. Die Eltern sind nicht immer selbst erkrankt, wenn sie neben einem veränderten Gen noch eine gesunde Kopie des betreffenden Gens haben. Auch Spontanmutationen sind möglich, bei denen Gene in einer der Keimzellen mutiert sind, ohne dass bei den Eltern diese Anomalie vorliegt. Man unterscheidet drei Arten von Erbkrankheiten: Chromosomenanomalien (veränderte Anzahl oder Struktur eines Chromosoms), polygenetische (Mutationen in mehreren Genen) und monogenetische Erbkrankheiten.
Monogenetische Erkrankungen werden durch eine Mutation in einem einzelnen Gen ausgelöst. Bei rezessiven Erkrankungen finden sich die Mutationen in beiden Kopien des Gens, während bei dominanten Erkrankungen die Mutation in einer Genkopie ausreicht für die Manifestation. Monogenetische Erkrankungen aufgrund einer Genmutation auf dem X-Chromosom können sowohl dominant (bei weiblichen Nachkommen) als auch rezessiv (bei männlichen Nachkommen) vererbt werden.
Die Kinder zeigen häufig schon früh Symptome. In vielen Fällen verläuft die Erkrankung schwerwiegend chronisch oder sogar tödlich. Obwohl die meisten Krankheiten außerordentlich selten vorkommen, ist insgesamt doch etwa 1 Prozent der Neugeborenen betroffen. Zu den häufigsten weltweit vorkommenden monogenetischen Krankheiten gehören Hämophilie, Mukoviszidose, Thalassämie und Phenylketonurie.
Mithilfe von molekularbiologischen Methoden konnte das mutierte Gen bei einem Drittel der bislang bekannten 6000 bis 10.000 monogenetischen Erkrankungen identifiziert werden. Häufig finden sich Punktmutationen oder Deletionen in verschiedenen Abschnitten des Gens. Daher existiert häufig eine große Zahl verschiedener Allel-Varianten, zum Beispiel bei Mukoviszidose mit mehr als 1000 Allel-Varianten des CFTR-Gens (1).
Bei der Bluterkrankheit oder Hämophilie werden aufgrund von Genmutationen die Gerinnungsfaktoren VIII oder IX nicht oder als Proteine mit einer reduzierten Aktivität produziert. Da diese Gene für Faktor VIII und IX auf dem X-Chromosom lokalisiert sind, betrifft die Hämophilie ausschließlich Männer. Etwa 80 Prozent der Patienten (1:4000 männliche Neugeborene) leiden an Hämophilie A, einem Faktor-VIII-Mangel. Seltener ist die Hämophilie B, ein Faktor-IX-Mangel (1:20.000 männliche Neugeborene).
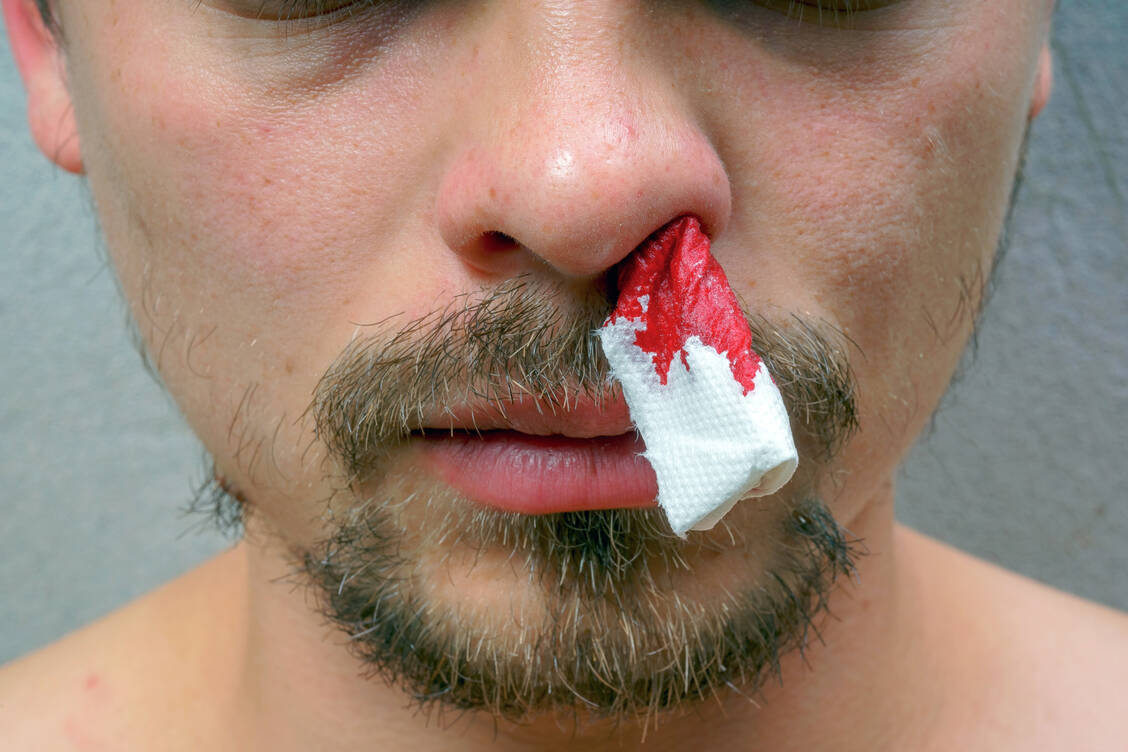
Patienten mit einer angeborenen Hämophilie leiden immer wieder an Blutungen. Gefährlicher als Nasenbluten sind Einblutungen in innere Organe und Strukturen, zum Beispiel in Gelenke, Muskeln, Gastrointestinaltrakt, Haut oder Gehirn. / Foto: Adobe Stock/Yashkin Ilya
Bei den Patienten treten Blutungen in verschiedenen Organen auf, zum Beispiel in Gelenken (Hämarthrosen), Muskeln, Gastrointestinaltrakt, Haut oder Gehirn. Besonders Blutungen in den Gelenken, die bis zur Zerstörung der betroffenen Gelenke führen können, beeinträchtigen die Lebensqualität der Patienten. Je nach Ausmaß des Traumas und der Plasmaspiegel von Faktor VIII oder Faktor IX kann es zu lebensbedrohlichen Blutungen kommen (2, 3).
Die aktuelle Therapie besteht in der lebenslangen Substitution der Gerinnungsfaktoren zur Kontrolle der Blutungen. Rekombinante Gerinnungsfaktoren sind im Gegensatz zu gereinigten Konzentraten sicher virusfrei.
Problematisch: Nach wiederholter Gabe entwickeln etwa 30 Prozent der Patienten mit schwerer Hämophilie A und 3 Prozent mit Hämophilie B Alloantikörper (Faktor-VIII- beziehungsweise Faktor-IX-Hemmkörper), wodurch die Wirksamkeit bei weiteren Infusionen sinkt (2).
Ein neuartiger Behandlungsansatz bei Hämophilie A ist die Gabe von Emicizumab (Hemlibra®). Der rekombinante humanisierte bispezifische monoklonale Antikörper bindet sowohl an aktivierten Faktor IX als auch an Faktor X und aktiviert dadurch Faktor X, was den Faktor VIII überflüssig macht (4). Das bedeutet: Emicizumab wirkt auch in Gegenwart von Hemmkörpern.
Der Antikörper ist zugelassen zur Routineprophylaxe von Blutungen bei allen Patienten mit Hämophilie A und Faktor-VIII-Hemmkörpern sowie bei schwerer Hämophilie A ohne Faktor-VIII-Hemmkörper. In klinischen Studien senkte Emicizumab bei beiden Patientengruppen signifikant und klinisch relevant die Blutungsrate um 80 bis 90 Prozent im Vergleich zur Standardtherapie, der Gabe von Gerinnungsfaktoren bei Bedarf (4). Emicizumab wird initial vier Wochen lang einmal wöchentlich subkutan gegeben; danach kann das Dosierungsintervall auf zwei oder vier Wochen verlängert werden. Schwerwiegende Nebenwirkungen sind thrombotische Mikroangiopathien und Thrombosen sowie oberflächliche Thrombophlebitiden, zum Teil mit Hautnekrosen.
In der klinischen Entwicklung befinden sich Concizumab, Fitusiran sowie mehrere gentherapeutische Ansätze.
Concizumab ist ein humanisierter monoklonaler Antikörper, der an TFPI (tissue factor pathway inhibitor) bindet und dessen Wirkung inhibiert. So kann Thrombin über den extrinsischen Weg mithilfe von Faktor Xa, TF (tissue factor) und Faktor VIIa produziert und damit das Faktor-VIII- und -IX-Defizit ausgeglichen werden. Studienergebnisse zeigen, dass die tägliche subkutane Gabe bei Hämophilie A und B mit und ohne Alloantikörper wirksam ist. Die jährliche Blutungsrate (ABR, annual bleeding rate) im Vergleich zur Gabe von Gerinnungsfaktoren nach Bedarf wird um circa 80 Prozent reduziert. Concizumab war gut verträglich. Am häufigsten wurden Nasopharyngitis, Reaktionen an der Injektionsstelle und obere Atemwegsinfektionen berichtet (5).
Mittlerweile wird Concizumab in Phase-III-Studien in einer größeren Patientengruppe untersucht (EXPLORER 6, 7 und 8). Im Frühjahr 2020 wurden die Studien aufgrund von thrombotischen Ereignissen bei drei Patienten unterbrochen, werden jedoch seit August 2021 nach Einführen weiterer Sicherheitsmaßnahmen fortgeführt (6).
Fitusiran, eine small interfering RNA (siRNA), bindet an die für Antithrombin kodierende mRNA, wodurch diese abgebaut wird und nicht mehr für die Proteintranslation zur Verfügung steht. Dadurch nimmt die Produktion des physiologischen Antikoagulans Antithrombin ab.
Die kürzlich vorgestellten Ergebnisse der Phase-III-Studien ATLAS A und B zeigten, dass die einmal monatliche subkutane Gabe von Fitusiran im Vergleich zur Gabe von Gerinnungsfaktoren bei Bedarf die jährliche Blutungsrate um 90 Prozent reduziert. Bei etwa der Hälfte der Studienteilnehmer traten keine behandlungsbedürftigen Blutungen mehr auf im Vergleich zu 5 Prozent in der Kontrollgruppe. Erhöhte Leberenzymwerte, obere Atemwegsinfektionen und Nasopharyngitis waren die häufigsten Nebenwirkungen. Circa ein Fünftel der Patienten hatte relevant erhöhte Leberenzymwerte, die über dem Dreifachen des oberen Grenzwerts lagen. Thrombotische Ereignisse wurden nicht berichtet (7).
Gentherapeutische Ansätze bei Hämophilie-Patienten verwenden fast ausschließlich in vivo verabreichte rekombinante nicht pathogene replikationsdefekte AAV-Vektoren (Kasten). Diese enthalten ein gesundes Gen für Faktor VIII oder IX sowie eine Hepatozyten-spezifische Promotorsequenz, die die Expression des episomalen Gens in den Leberzellen ermöglicht, da sowohl Faktor VIII als auch Faktor IX physiologisch in der Leber synthetisiert werden. Mehrere Gentherapien werden derzeit klinisch erprobt (9).
Bei Patienten mit Hämophilie A ist die klinische Testung von Valoctocogen roxaparvovec (AAV5-hFVIII-SQ, Valrox®) am weitesten fortgeschritten (Tabelle). Das Transgen ist eine komplementäre einzelsträngige DNA des Faktors VIII, der die B-Domäne fehlt, was die biologische Aktivität nicht vermindert (hFVIII-SQ). Das produzierte Protein hat dieselbe Aminosäuresequenz wie rekombinante Gerinnungsfaktoren, die kein erhöhtes Risiko für Alloantikörper zeigen (Beispiel: ReFacto®).
| Name, Firma | Transgen | Vektor | Klinische Entwicklung |
|---|---|---|---|
| Hämophilie A | |||
| Valoctocogen roxaparvovec (Valrox®), Biomarin | ssFVIII-SQ | AAV5 | Phase III abgeschlossen, Zulassungsantrag eingereicht |
| Giroctocogen fitelparovec, Sangamo/Pfizer | ssFVIII-SQ | AAV2/6 | Phase III |
| SPK-8011, Spark | ssFVIII-SQ | modifizierter AAV3 | Phase II |
| BAY2599023 (DTX201), Bayer/Ultragenyx | ssFVIII-SQ | AAVhu37 | Phase I/II |
| BAX 888, Baxalta/Shire | ssFVIII-SQ | AAV8 | Phase I/II |
| AAV2/8-HLP-FVIII-V3, University College of London | ssFVIII-V3 | AAV2/8 | Phase I |
| CD68-ET3-LV CD34+, Expression Therapeutics | ET3 | Lentivirus | Phase I |
| Hämophilie B | |||
| Etranacogen dezaparvovec, UniQure, CSL Behring | ssFIX-R338L | AAV5 | Phase III |
| Fidanacogen elaparvovec (RAAV-SPARK100-HFIX-PADUA), Pfizer/Spark | ssFIX-R338L | SPK100 | Phase III |
| Verbrinacogen setparvovec, Freeline | FLT180a FIX Padua | AAVS3 | Phase I/II |
In der Phase-III-Studie GENE 8-1 mit 134 Patienten reduzierte eine einmalige Infusion die jährliche Blutungsrate (ABR) um 84 Prozent im Vergleich zur Gabe von Faktor-VIII-Konzentrat und den Bedarf an Faktor VIII um 99 Prozent. Nach zwei Jahren war die produzierte Menge an Faktor VIII geringer, reichte jedoch noch aus für eine hämostatische Wirksamkeit (ABR 0,9). Die Gentherapie wurde gut vertragen; thromboembolische Nebenwirkungen traten nicht auf und es wurden keine Antikörper gegen Faktor VIII gebildet. Erhöhte Leberenzymwerte waren die häufigste Nebenwirkung, jedoch vorübergehend und ohne klinische Symptome. Bei den Zulassungsbehörden in den USA und in Europa (FDA und EMA) sind Zulassungsanträge eingereicht (10).
In einer sehr frühen klinischen Erprobungsphase befindet sich ein gentherapeutischer Ex-vivo-Ansatz. Dabei werden dem Patienten CD34+-Stammzellen entnommen. Mithilfe eines Monozyten-spezifischen Lentivirus wird ex vivo ein hochaktives Faktor-VIII-Gen eingeschleust. Der Patient erhält eine Chemotherapie mit Busulfan und danach die modifizierten Stammzellen als Infusion. Mit Studienergebnissen wird frühestens 2025 gerechnet (11).
Auch für Hämophilie B sind mehrere Gentherapien in der klinischen Erprobung. Am weitesten fortgeschritten ist Etranacogen dezaparvovec (Tabelle). Das verwendete Transgen ist ein physiologisch vorkommendes mutiertes Faktor-IX-Gen. Die Missense-Mutation erhöht die Aktivität des Proteins um das Achtfache im Vergleich zum Wildtyp-Faktor IX.
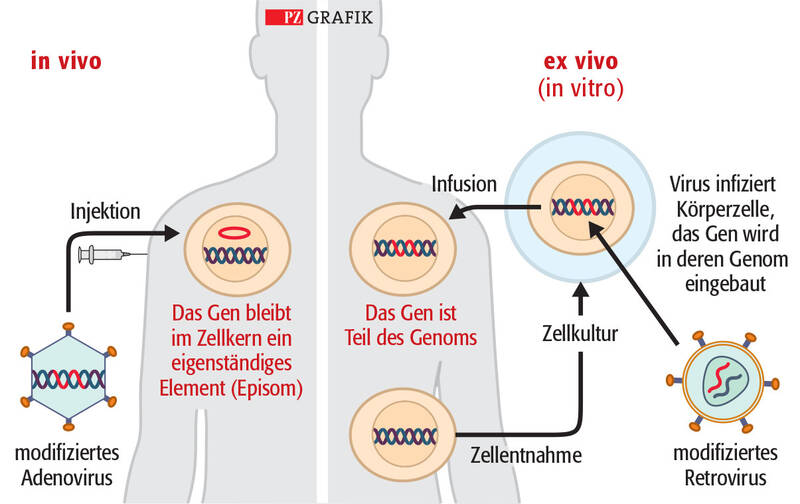
Foto: PZ/Stephan Spitzer
Um genetische Information in die Zielzellen einzuschleusen, werden virale Vektoren verwendet. Diese sogenannte Transduktion kann ex vivo oder in vivo erfolgen. Für eine Gentherapie werden zwei Arten von Viren eingesetzt: Retroviren wie Lentiviren beziehungsweise Adeno- oder Herpesviren.
Um ein Virus als Vektor einzusetzen, muss es zunächst genetisch so modifiziert werden, dass nach Integration des therapeutischen Gens keine Viruspartikel gebildet werden, also das Virus sich nicht repliziert. Zu diesem Zweck werden die entsprechenden viralen Abschnitte des Erbmaterials entfernt.
Bei der Ex-vivo-Gentherapie werden dem Patienten die Zielzellen zunächst entnommen. Mithilfe von Lentiviren, die sich nicht vermehren können, wird das therapeutische Gen stabil in die entnommenen Zellen integriert; das heißt, dass das Gen bei der Zellteilung an die Tochterzellen weitergegeben wird. Anschließend werden die in Zellkultur vermehrten modifizierten Zellen dem Patienten wieder verabreicht. Das Verfahren ist für Zellen geeignet, die sich relativ leicht aus dem Körper gewinnen lassen, zum Beispiel Stammzellen aus dem Knochenmark, und die sich anschließend replizieren.
Bei einer In-vivo-Gentherapie werden Viren, die das therapeutische Gen enthalten, direkt in den Körper des Patienten infundiert oder injiziert. Derzeit werden vor allem Adeno-assoziierte Viren (AAV) als Genfähren verwendet. Die AAV-Vektoren integrieren ihr Erbgut nicht in das Genom der menschlichen Zelle, sondern es verbleibt als sogenanntes Episom im Zellkern und wird während einer Zellteilung daher nicht vermehrt. AAV-Vektoren werden deshalb für das Einbringen von Genen in postmitotisches Gewebe eingesetzt, das sich nicht vermehrt, zum Beispiel Retina, Leber oder Muskel. Die Selektivität der Viren für bestimmte Gewebe wird durch modifizierte Virushüllen und gewebespezifische Promotoren verbessert (8).
In der Phase-III-Studie HOPE-B erhielten 54 Patienten mit schwerer Hämophilie B zunächst sechs Monate ihre Standardtherapie, danach einmal eine Infusion Etranacogen dezaparvovec. Zwölf Monate nach der Infusion betrug die Blutungsrate 1,5; im Vergleich dazu lag die Blutungsrate bei 4,2 während der ersten sechs Monate unter Standardtherapie. Auch 18 Monate nach der Infusion hatten die Patienten eine klinisch relevante Faktor-IX-Aktivität. Die Gentherapie wurde gut vertragen; rund 80 Prozent der Nebenwirkungen waren mild. Faktor-IX-Antikörper traten nicht auf. Die Zulassungsunterlagen sollen 2022 bei den Behörden eingereicht werden (12).
Auch für Fidanacogen elaparvovec liegen ermutigende Ergebnisse einer Phase-II-Studie mit zehn Männern vor. Mithilfe eines Adeno-assoziierten Vektors wurde ein hochaktives Faktor-IX-Transgen infundiert. Die jährliche Blutungsrate sank von 11,1 auf 0,4 nach der Infusion. Die Faktor-IX-Aktivität lag bei 30 Prozent. Während der 18-monatigen Nachbeobachtungszeit traten bei neun der zehn Probanden keine Blutungen auf; acht benötigten keine Gerinnungsfaktoren und die anderen beiden nur in geringem Maß. Die Therapie wurde gut vertragen. Weder Thrombosen oder erhöhte Laborwerte noch gegen Faktor IX gerichtete Antikörper wurden beobachtet (13). Die zulassungsrelevanten Studien zur Langzeitsicherheit und -wirksamkeit dauern noch an.
Die zystische Fibrose (CF) oder Mukoviszidose ist eine autosomal-rezessiv vererbte Stoffwechselstörung. Die Inzidenz liegt bei 1 zu 2500 Neugeborenen in Europa. In Deutschland leben schätzungsweise 6000 Betroffene. Man kennt mittlerweile rund 2000 Mutationen, die zu einer unterschiedlichen Ausprägung der Erkrankung führen können. Dabei kann der Patient auch zwei unterschiedliche Mutationen auf den langen Arm des Chromosoms 7 haben.
Die Therapie der Mukoviszidose ist umfangreich, aber viele Patienten können damit erwachsen werden. Eine heilende Gentherapie ist nicht in Sicht. / Foto: Adobe Stock/Dragana Gordic
Die häufigste Mutation, F508del, liegt bei rund 90 Prozent der Mukoviszidose-Patienten vor. Dabei fehlt dem Cystic Fibrosis Transmembrane Regulator-(CFTR-)Protein, das den Chlorid-Ionenkanal bildet, ein Phenylalanin in Position 508. Das dadurch fehlgefaltete Protein wird entweder zu wenig oder funktionell eingeschränkt an der Zelloberfläche exprimiert. In der Folge sinkt die Konzentration der osmotisch wirksamen Chlorid-Ionen im Sekret verschiedener Drüsen. Das hochvisköse Sekret führt unter anderem zu stark verklebten Atemwegen und einer Malabsorption im Verdauungstrakt.
Die Standardtherapie der Patienten beinhaltet die Inhalation von Betasympathomimetika und einer gentechnisch hergestellten DNase. Zudem erhalten die Patienten Verdauungsenzyme, Antibiotika zur Behandlung von immer wieder auftretenden Atemwegsinfektionen und eine hochkalorische Ernährung (14, 15).
Sogenannte CFTR-Modulatoren haben die Therapie der CF wesentlich verbessert. Mittlerweile sind vier Arzneistoffe zugelassen. Der CFTR-Potenziator Ivacaftor (Kalydeco®) erhöht bei Patienten mit G551D-Mutation die Wahrscheinlichkeit, dass der defekte Chlorid-Ionenkanal geöffnet wird. Die anderen drei wirken als CFTR-Korrektoren und erhöhen die Menge an funktionellem CFTR an der Zelloberfläche. Lumacaftor (Orkambi®) und Tezacaftor/Ivacaftor (Symkevi®) sind für Patienten mit F508del-Mutation zugelassen. Für diese Mutation plus eine weitere Mutation in einem anderen Allel wurde die Dreifachkombination von Ivacaftor/Tezacaftor/Elexacaftor (Kaftrio®) im August 2020 in Europa zugelassen.
Die Wirksamkeit der Dreierkombination wurde in zwei klinischen Studien belegt. In beiden Studien konnte die neue Therapie das vorhergesagte forcierte exspiratorische Ein-Sekunden-Volumen (ein etablierter Marker in der Mukoviszidose-Behandlung) deutlich verbessern: um 13,8 Prozent gegenüber Placebo in der ersten Studie sowie um 10 Prozent im Vergleich zur Zweierkombination. Weitere positive Effekte waren eine Zunahme des Body-Mass-Indexes – CF-Patienten sind aufgrund von Malabsorptionsstörungen oft untergewichtig – sowie weniger pulmonale Exazerbationen. Das Sicherheitsprofil entsprach dem der Zweierkombination. Als häufigste Nebenwirkungen traten Kopfschmerzen, Infekte der oberen Atemwege, Bauchschmerzen, Durchfall, Hautausschläge und erhöhte Leberenzymwerte auf (16, 17).
Auch bei CF-Patienten wurden verschiedene gentherapeutische Ansätze klinisch erprobt, die vor allem das Ziel hatten, korrekte Kopien des CFTR-Gens in die Atemwege einzuschleusen. Jedoch waren bisherige Versuche mit AAV- oder Lentivirus-Vektoren wenig erfolgreich. Da sich die Epithelzellen in den Atemwegen ständig erneuern, kommt es nur zu einer vorübergehenden Genexpression. Weiterhin verhindert der Schleim in den Atemwegen ein effizientes Einschleusen in die Zellen. Die Mehrfachbehandlung löste starke Immunreaktionen aus, die die Wirksamkeit verringerten (18).
Bisherige Versuche mit einem nicht viralen Liposomvehikel zeigten ebenfalls nur eine begrenzte Wirksamkeit. Die Ein-Sekunden-Kapazität der Lunge verbesserte sich klinisch gering, wenn auch signifikant um 3,7 Prozent gegenüber Placebo. Weiterhin stabilisierte sich die Lungenfunktion, während sie in der Placebogruppe abnahm (19). Für eine effektivere Transduktion der Zielzellen ist die Entwicklung neuer Vektoren notwendig (18).
Die häufigste monogenetische Erkrankung vor allem in ehemaligen Malariagebieten im Mittelmeerraum, im Vorderen Orient sowie in Afrika und Asien ist die Thalassämie. Etwa 60.000 Neugeborene kommen jedes Jahr mit einer schweren Form der Erkrankung auf die Welt. In Deutschland ist die Erkrankung mit circa 500 Patienten mit einer schwerwiegenden Form sehr selten. Ursache ist eine Mutation oder Deletion in den Genen für die α- oder β-Untereinheit des Hämoglobin, wodurch kein funktionsfähiges Hämoglobin in den Erythrozyten produziert wird (20, 21).
Bei der α-Thalassämie befindet sich der Defekt auf Chromosom 16, das für die α-Untereinheit kodiert. Der Schweregrad der Erkrankung hängt davon ab, wie viele Allele betroffen sind. Bei der schwersten Form sind alle vier Allele des α-Gens inaktiv; unbehandelt stirbt der Embryo bereits intrauterin. Wenn drei Allele inaktiv sind, liegt die sogenannte HbH-Krankheit vor. Dies ist eine leichtere Form der Thalassämie, bei der die Patienten eine hämolytische Anämie aufweisen sowie Leber und Milz vergrößert sind, viele jedoch klinisch kaum Symptome haben und selten Bluttransfusionen benötigen, da noch Hämoglobin gebildet werden kann. Im Erwachsenenalter können Komplikationen wie kardiale Probleme, Gallensteine, Unterschenkelgeschwüre und Folsäuremangel auftreten.
Die β-Thalassämie ist die häufigere Form. Verantwortlich sind Mutationen des Gens auf dem Chromosom 11, das die β-Untereinheit des Hämoglobins kodiert. Bekannt sind mehr als 4000 verschiedene Mutationen, die meist autosomal-rezessiv vererbt werden.
Bei der Thalassämie minor ist nur ein Genallel betroffen und die Patienten haben meist keine klinischen Symptome außer einer leicht vergrößerten Milz. Diese Form ist nicht behandlungsbedürftig.
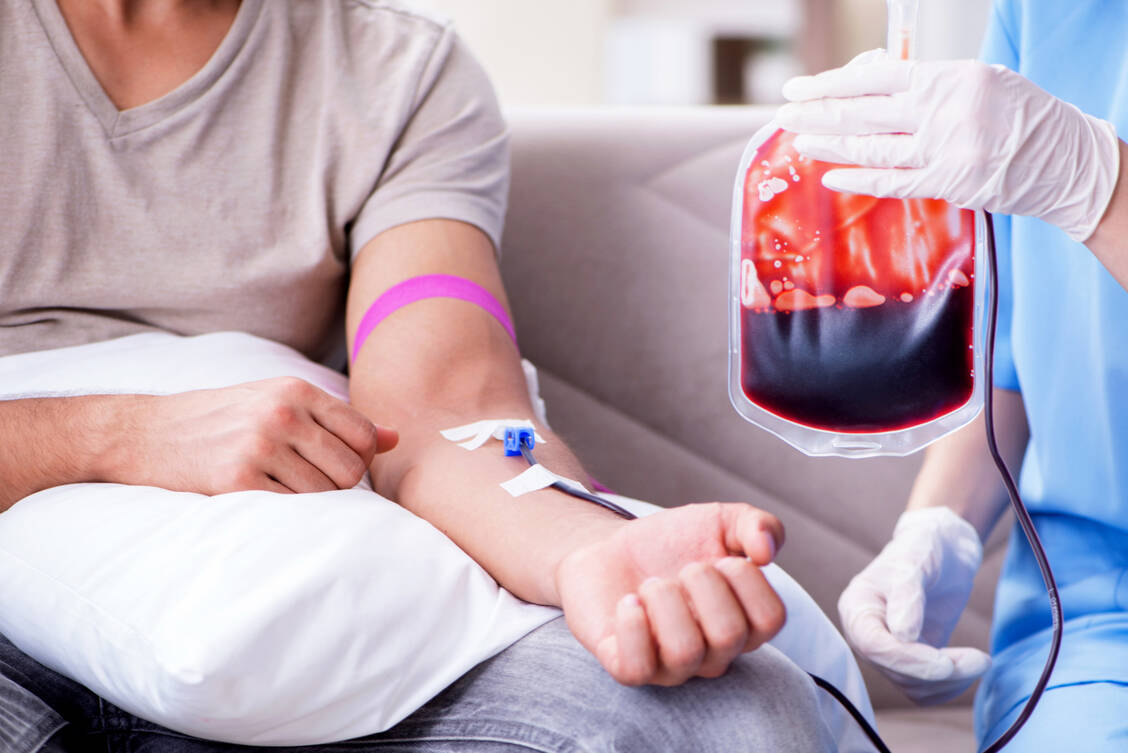
Patienten mit Thalassämien können kein funktionsfähiges Hämoglobin bilden. Je nach Schweregrad der Erkrankung brauchen sie regelmäßige Infusionen von Erythrozytenkonzentraten. / Foto: Shutterstock/Elnur
Bei der Thalassämie major, bei der beide Allele betroffen sind, werden die β-Globinketten nicht synthetisiert und es kann kein normales HbA1 (α2β2) produziert werden. Durch den starken Überschuss an α-Globinen entstehen defekte instabile Erythrozyten, die bereits im Knochenmark zugrunde gehen. Bereits wenige Monate nach der Geburt vergrößern sich Leber und Milz stark. Im weiteren Verlauf treten Wachstumsstörungen, schwere Schäden innerer Organe und Knochenfehlbildungen auf. Durch den Mangel an Erythrozyten und Hämoglobin kann Sauerstoff nicht effektiv transportiert werden: Eine schwere Anämie ist die Folge. Unbehandelt führt die Erkrankung im frühen Kindesalter zum Tod (20, 21).
Standardbehandlung bei β-Thalassämie major ist die Gabe von Erythrozytenkonzentraten sowie eine begleitende Chelattherapie (Deferasirox oder Dexferoxamin), um eine Eisenüberladung zu verhindern, die zu Organschäden, zum Beispiel am Herzen, führen kann. Gibt es einen HLA-identischen Spender, beispielsweise ein Geschwisterkind, ist eine hämatopoetische Stammzelltransplantation die Therapie der Wahl (20).
Mitte 2019 wurde mit Betibeglogene autotemcel (Zynteglo®) eine Gentherapie, die das Erbgut von Blutstammzellen verändert, für Patienten mit β-Thalassämie von der EMA zugelassen (22). Die Patienten werden zunächst mit Granulozyten-Kolonie-stimulierendem Faktor (G-CSF) und Perixafor, einem Chemokinrezeptor-CXCR4-Antagonisten, behandelt, um die CD34+-Stammzellen zu mobilisieren. Dabei werden die Stammzellen aus dem Knochenmark in die Blutbahn freigesetzt, sodass sie mittels Apherese entnommen werden können. Im Labor werden die Zellen mithilfe eines nicht replikationsfähigen Lentivirus-Vektors, der eine funktionstüchtige Genvariante enthält, transfiziert.
Vor der Infusion der modifizierten Stammzellen erhalten die Patienten eine myeloablative (knochenmarkszerstörende) Chemotherapie mit Busulfan. Die modifizierten Stammzellen siedeln sich im Knochenmark an und differenzieren zu Erythrozyten, die biologisch aktives β-Globin-Protein produzieren, das zusammen mit α-Globin funktionelles Hämoglobin bildet.
Die Behandlung ist zugelassen für Patienten ab zwölf Jahren, die an einer transfusionsabhängigen Form von β-Thalassämie (TDT) leiden, keinen passenden Spender für eine Stammzelltransplantation haben und die noch β-Globin-Protein produzieren können.
Betibeglogene autotemcel wurde bislang in vier klinischen Studien und einer Langzeitstudie an 32 erwachsenen und jugendlichen TDT-Patienten untersucht. In der Hauptstudie benötigten nach einmaliger intravenöser Infusion elf von 14 Patienten (79 Prozent) keine Erythrozyten-Transfusionen mehr. Das heißt: Innerhalb der zwei Studienjahre lag ihr Hämoglobinwert über einen Zeitraum von zwölf Monaten mindestens bei 9 g/dl oder höher. Ob die Wirkung der Gentherapie lebenslang anhält, kann man noch nicht beurteilen. Bislang liegen Daten für einen Beobachtungszeitraum von fünf Jahren vor.
Die Behandlung wurde im Allgemeinen gut vertragen. In den klinischen Studien mit insgesamt 48 Patienten traten Thrombozytopenien, Bauch- und Brustschmerzen, Schmerzen in den Extremitäten, Atemnot und Rötungen der Haut auf (22). Einen Zusammenhang mit einer akuten myeloischen Leukämie bei zwei Patienten in einer anderen Studie, bei der der gleiche virale Vektor verwendet wurde, hat die europäische Zulassungsbehörde nach eingehender Prüfung ausgeschlossen. Jedoch kann nicht ausgeschlossen werden, dass die chemotherapeutische Vorbehandlung das Krebsrisiko erhöht. Die Patienten sollen deshalb 15 Jahre lang mindestens einmal jährlich auf Zeichen von Blutkrebs untersucht werden (23).
Codon: Nukleotidtriplett, das für eine Aminosäure kodiert
Deletion: Mutation, bei der es zum Verlust von Nukleotiden oder DNA-Sequenzen kommt
Episom: eigenständiges genetisches Element außerhalb des Genoms
Insertionsmutation: Einbau von zusätzlichen Nukleotiden oder DNA-Sequenzen in eine DNA-Sequenz. Eine Insertion führt zu einer Leserasterverschiebung, das Genprodukt, das auf dem Gen codiert ist, wird nicht mehr korrekt hergestellt.
Missense-Mutation: Punktmutation, bei der eine einzelne Nukleotidänderung zu einem Codon führt, das für eine andere Aminosäure kodiert
Promotor: Nukleotid-Sequenz der DNA, die die Expression eines Gens ermöglicht, indem sie an die RNA-Polymerase und weitere Proteine, sogenannte Transkriptionsfaktoren, bindet, die den Start des »Ablesens« des Gens durch die RNA-Polymerase vermitteln
Transduktion: Einbringen des genmodifizierten Materials mithilfe viraler Vektoren in eine Zielzelle
Transkription: Synthese von RNA durch Ablesen einer DNA
Translation: Synthese von Proteinen im Anschluss an die Transkription
Störungen des Phenylalanin-Stoffwechsels können zu abnorm erhöhten Konzentrationen der Aminosäure Phenylalanin im Blut führen. Auslöser der Hyperphenylalaninämie (HPA) sind zwei angeborene Stoffwechselstörungen: die Phenylketonurie (PKU) sowie der Mangel an Tetrahydrobiopterin (BH4) mit schätzungsweise rund 35.000 Patienten in Europa. Dabei ist der BH4-Mangel mit 1 bis 2 Prozent aller Fälle äußerst selten.
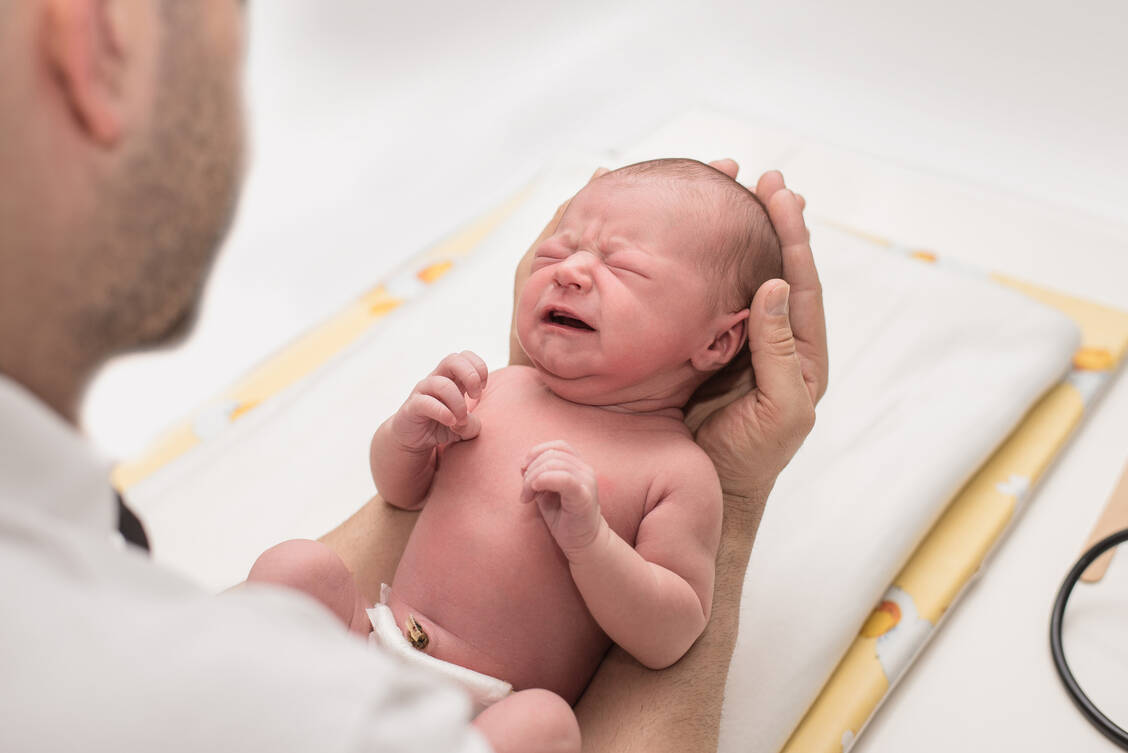
Neugeborene Babys werden heute gründlich untersucht. Beim Screening wird auch auf erhöhte Phenylalanin-Blutspiegel getestet. / Foto: Adobe Stock/S.Kobold
Ursache der PKU ist ein Mangel des Enzyms Phenylalanin-Hydroxylase (PAH) durch eine Mutation des Gens auf Chromosom 12. PAH wird für den Stoffwechsel von Phenylalanin benötigt. Ist das aktive Enzym nicht ausreichend vorhanden, so steigt nach Nahrungsaufnahme der Phenylalanin-Spiegel im Blut und Gehirn (Normbereich 50 bis 120 μmol/l) auf abnorm hohe Werte. Bereits bei Konzentrationen über 600 µMol/l kann es zu Komplikationen wie schweren Entwicklungsverzögerungen, Gehirnschäden, Krämpfen, Muskelzittern und Wahrnehmungsstörungen kommen. In fast allen westlichen Industrienationen wird beim Neugeborenen-Screening auf erhöhte Phenylalanin-Blutspiegel getestet.
Bei einem Mangel des Cofaktors BH4 wird Phenylalanin unzureichend zu Tyrosin umgewandelt. Da BH4 auch ein Cofaktor für die Enzyme Tyrosin-Hydroxylase und Tryptophan-Hydroxylase ist, ist bei einem Mangel auch kein normaler Metabolismus der Aminosäuren Tyrosin und Tryptophan möglich. In der Folge sind die Konzentrationen an Neurotransmittern wie Dopamin und Serotonin verringert, was zu extrapyramidalen Bewegungsstörungen, Temperaturregulationsstörungen und verändertem Muskeltonus führt (24).
Standardbehandlung ist eine restriktive Phenylalanin-arme Diät. Sapropterin, eine synthetische Form des natürlich vorkommenden 6R-Tetrahydrobiopterins (6R-BH4), wird zur Behandlung der Hyperphenylalaninämie eingesetzt. Es erhöht die Aktivität der fehlerhaften Phenylalanin-Hydroxylase und steigert dadurch den oxidativen Metabolismus von Phenylalanin. Dessen Konzentration im Blut wird gesenkt beziehungsweise werden die Spiegel im Normbereich gehalten und die Toleranz gegenüber einer diätetischen Phenylalanin-Zufuhr gesteigert. Bei Patienten mit einem BH4-Mangel gleicht Sapropterin die zu niedrigen BH4-Spiegel aus und normalisiert die Aktivität des PAH-Enzyms (24).
Bei der PKU sind zwei gentherapeutische Ansätze in der frühen klinischen Erprobung. Bei beiden Therapien wird ein nicht pathogener replikationsdefekter AAV-Vektor verwendet, der ein intaktes humanes PAH-Gen enthält, sowie eine Hepatozyten-spezifische Promotorsequenz, die die Expression des episomalen Gens in den Leberzellen ermöglicht. Durch eine einmalige intravenöse Infusion soll PAH synthetisiert und damit der Phenylalanin-Metabolismus normalisiert werden (25, 26). Ergebnisse liegen bislang nicht vor. Jedoch werden derzeit keine Patienten mit BMN-307 behandelt, nachdem bei Mäusen nach Gabe von hohen Dosen Tumore in der Leber auftraten (27).
Mittlerweile gibt es gentherapeutische Ansätze für eine Reihe von monogenetischen Erkrankungen. Studien mit meist wenigen Patienten machen Hoffnung auf eine längerfristige Wirksamkeit. Jedoch sind noch viele Fragen zum optimalen Zeitpunkt der Behandlung, zur Langzeitwirkung und Sicherheit der Gentherapien ungeklärt. Für einige Erkrankungen, zum Beispiel CF, sind die bislang verwendeten Vektoren nicht effizient genug sind, um das Gen stabil einzuschleusen, und die mehrfache Gabe löst Immunreaktionen aus. Auch die hohen Kosten für die Gentherapien werfen Fragen auf.
Bettina Wick-Urban studierte Pharmazie an der Albert-Ludwigs-Universität in Freiburg. Nach ihrer Promotion in Basel und Freiburg mit einer molekularbiologischen Arbeit im Bereich der experimentellen Onkologie arbeitete sie zunächst als Referentin bei der Arzneimittelinformationsstelle der ABDA. Danach wechselte sie in die pharmazeutische Industrie, wo sie seitdem in verschiedenen Positionen in der klinischen Forschung, Arzneimittelsicherheit und Medizin tätig ist, davon zwei Jahre in den USA. Zwischenzeitlich schloss sie ein Journalismusstudium ab.


