 |  | Hans-Peter Lipp |
 |
09.05.2021 08:00 Uhr |
Verliert ein Wirkstoff innerhalb der EU seinen Patentschutz, so ergibt sich bei den meisten Substanzen zunächst die Frage, ob es sich um eine chemisch genau definierbare Einzelsubstanz oder um ein rekombinant gewonnenes (Glyko)protein (Biological) handelt.
Im ersten Fall befindet man sich auf regulatorischer Ebene im Bereich der Generika, im letzteren Fall im Bereich der Biosimilars, deren Zulassung innerhalb der EU ausschließlich zentral über die European Medicines Agency (EMA) erfolgen kann (Grafik). Eine Sonderstellung nehmen in diesem Zusammenhang die niedermolekularen Heparine (NMH) ein, da sie regulatorisch den Biosimilars zugeordnet werden, selbst aber keine Glykoproteine, sondern partialsynthetisch gewonnene Folgeprodukte des unfraktionierten Heparins darstellen und damit biologischen Ursprungs sind. Im Gegensatz zu den rekombinanten Produkten erfordert die Zulassung eines NMH-Biosimilars allerdings nicht zwingend eine Phase-III-Studie im direkten Vergleich mit einem Referenzarzneimittel in einem klinisch relevanten Anwendungsgebiet (23).
Auch wenn mit dieser Kategorisierung die Zulassung eines Großteils von Arzneimitteln nach Patenschutzverlust klar geregelt ist, ergeben sich insbesondere für die Stoffklasse der NBCD noch eine Reihe offener Fragen, da ihre substanzspezifischen Charakteristika im Rahmen der genannten etablierten Zulassungsprozesse für Generika bisher nicht ausreichend berücksichtigt werden.
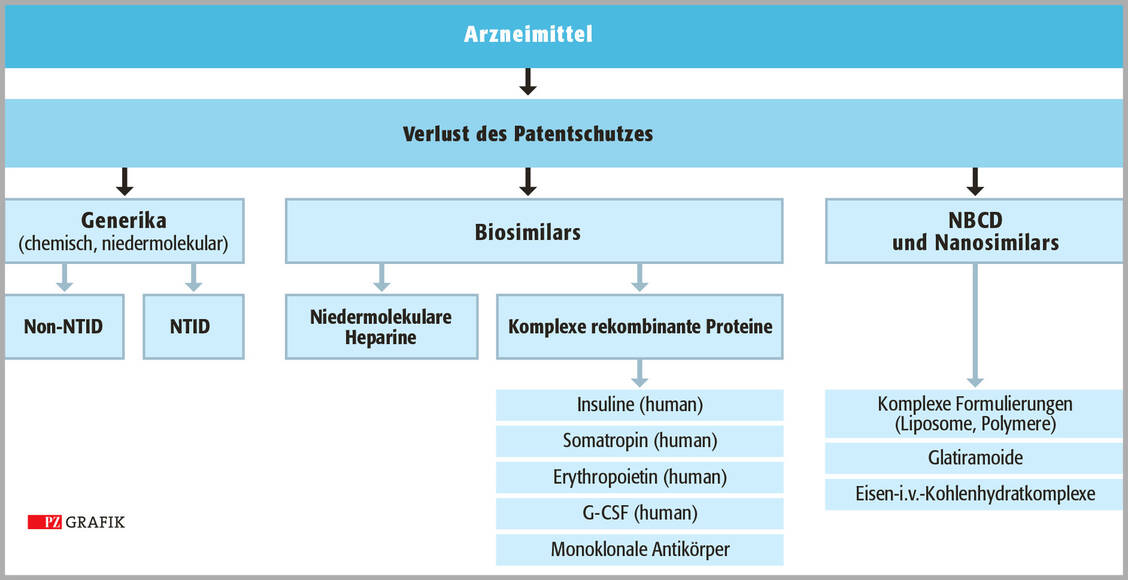
Die derzeit diskutierte Einteilung und Unterscheidung von Arzneimitteln nach Patentschutzverlust in Generika, Biosimilars und NBCD beziehungsweise NBCD-Similars. NTID = Generika mit geringer therapeutischer Breite (mod. nach 28) / Foto: PZ/Stephan Spitzer
Wird in diesem Zusammenhang häufig auch der Begriff Nanopharmazeutika zur weitergehenden Produktbeschreibung der NBCD verwendet, so weisen ihre hinsichtlich der pharmakologischen Aktivität bedeutsamen Nanopartikel in der Summe ihrer inneren und äußeren Dimension eine Größe zwischen 1 nm und 100 nm auf und liegen teilweise in einem wässrigen Medium in kolloidal gelöster Form vor. Dabei trägt die Nanodimension zu den pharmakologischen Effekten des NBCD bei (1–5).
Wird ein Fertigarzneimittel innerhalb der EU zugelassen, so entscheidet zunächst die Art des Wirkstoffs, inwieweit eine zentrale Zulassung über die EMA erfolgen muss. Hierzu zählen sämtliche rekombinant hergestellten Arzneimittel, alle Gen- beziehungsweise Zelltherapeutika, Orphan Drugs, Virustatika und Humanarzneimittel, die einen innovativen Ansatz zur Behandlung von Krebserkrankungen, Diabetes mellitus, neurodegenerativen oder autoimmun-bedingten Erkrankungen bieten. Für die rekombinant gewonnenen Wirkstoffe spielt es damit keine Rolle, ob es sich um Produkte eines Erst- oder Zweitanbieters (Biosimilar) handelt.
»The process makes the product«: Zwar gelten auch für NBCD die Zulassungsbedingungen ähnlich denen der Biologicals und Biosimilars. Die Zulassung für das Produkt eines Zweitanbieters kann aber bisher dezentral auf rein nationaler Ebene erfolgen, da die NBCD per se bisher nicht Gegenstand eines verpflichtend zentralen Zulassungsprozesses sind, es sei denn, die Art des Wirkstoffs macht dies erforderlich (1–5, 24).

Verliert ein Wirkstoff innerhalb der EU seinen Patentschutz, so ergibt sich bei den meisten Substanzen zunächst die Fragestellung, ob es sich um eine chemisch genau definierbare Einzelsubstanz oder um ein rekombinant gewonnenes (Glyko)protein handelt. / Foto: Adobe Stock/Grecaud Paul
Um es noch einmal zusammenzufassen: Das NBCD Glatiramer ist als Ergebnis eines komplexen synthetischen Herstellungsverfahrens zu verstehen, wobei die einzelnen Herstellungsschritte am Ende entscheidenden Einfluss auf die genaue Zusammensetzung des Endprodukts haben.
Eine Bioäquivalenz-Prüfung wie bei einer Generika-Zulassung ist nicht durchführbar, da die einzelnen Polypeptide im Plasma nicht erfasst werden können. Der Nachweis einer Immunmodulation auf der Basis von Surrogatparametern bei gesunden Probanden hätte wiederum eine zu ungenaue Aussagekraft für die Anwendung bei Multipler Sklerose (MS) zur Folge gehabt.
Damit wurde letztendlich der Weg für eine Hybridzulassung geebnet; in diesem Fall wurde – im Gegensatz zu einer Generika-Zulassung – eine Phase-III-Vergleichsstudie gegenüber dem Referenzarzneimittel Copaxone® für die Zulassung des Nachfolgepräparats (zum Beispiel Clift®) von der EMA verpflichtend vorgeschrieben, ähnlich wie bei den Biosimilars (7–9, 25).
Der zentrale Eisen-III-Oxid-Hydroxid-Kern bei Eisen-III-Kohlenhydratkomplexen ist bei den Präparaten der dritten Generation fest in die jeweiligen Kohlenhydratstrukturen eingebettet, sodass am Ende eine hochkomplexe nanokolloidale Struktur entsteht, die über konventionelle analytische Methoden nicht mehr ausreichend genau präparatespezifisch charakterisiert werden kann. Hinzu kommt eine gewisse Mikroheterogenität des Endprodukts von Charge zu Charge – ähnlich wie bei den Biologicals und Biosimilars (1–5).
Bisher können Produkte von Zweitanbietern, zum Beispiel Fe(III)-Sucrose, im Gegensatz zu Biosimilars national zugelassen werden. Da eine Bioäquivalenz-Prüfung im Rahmen der verwendeten Komplexe im Plasma analytisch nicht möglich ist, müssen zwangsläufig Surrogatparameter wie zum Beispiel ∆TSAT% oder ∆Hb für vergleichende Untersuchungen herangezogen werden. Erfahrungen aus der Praxis haben allerdings gezeigt, dass im Rahmen eines Aut-idem-Wechsels von einem Referenzarzneimittel (Venofer®) auf ein ISS Probleme in der Verträglichkeit und Wirksamkeit auftraten (26).
Experten fordern deshalb seit Längerem, dass nach einem Patenschutzverlust von parenteralen Eisen-III-Kohlenhydratkomplexen Produkte von Zweitanbietern ebenfalls eine Hybridzulassung – ähnlich wie bei Glatiramer – durchlaufen müssen, um die klinische Nicht-Unterlegenheit des Nanopharmazeutikums gegenüber dem Referenzarzneimittel fundierter zu belegen.
Diese Forderung ist auch im Einklang mit dem »Reflection Paper« des »Committee for Medicinal Products for Human Use« (CHMP) der EMA, das physikalisch-chemische Produktcharakterisierungen als Nachweis für eine möglichst hohe Übereinstimmung mit bereits zugelassenen Präparaten unter evidenzbasierten Aspekten als nicht ausreichend betrachtet (1–5).
Ob damit ein substanzspezifisch unterschiedliches Risiko für schwere HSR Grad ≥ 3 tatsächlich herausgearbeitet werden kann, bleibt offen, da sicher eine relativ hohe Zahl an Patienten notwendig sein dürfte, um eine signifikante Differenz zwischen zwei Behandlungsgruppen herausarbeiten zu können.
Hingegen lassen sich unterschiedliche Inzidenzen an Grad ≥ 1 HSR zweifelsohne relativ gut erfassen (11, 14). Die klinische Bedeutung der HSR darf in diesem Zusammenhang nicht unterschätzt werden, da das Auftreten einer schweren lebensbedrohlichen Form keine weitere Anwendung eines intravenös zu applizierenden Eisen-III-Kohlenhydratkomplexes jedweder Art erlaubt (14).
Sehr komplex sind zweifelsohne auch die parenteral zu applizierenden liposomalen Einbettungen mit Amphotericin B, Anthrazyklinen oder Irinotecan. Obwohl für einige Präparate das Patent schon seit Längerem abgelaufen ist, stehen bis heute keine entsprechenden Nanosimilars, also Produkte von Zweitanbietern, innerhalb der EU zur Verfügung.
Dieses dürfte auch mit ihrer Komplexität und notwendigen Chargenkonformität in Zusammenhang stehen. Da dem frei verfügbaren, prozentualen Wirkstoffanteil eine maßgebliche Bedeutung im Toxizitätsprofil der Formulierung zukommt, sind auch in diesem Fall hohe Ansprüche an das Zulassungsverfahren für Zweitanbieter zu stellen (18).
Hingegen wird man – trotz ebenfalls hoher Wirkstoffkomplexität – nicht dieselben Ansprüche an nicht resorbierbare NBCD wie zum Beispiel Sevelamer stellen, da ihre systemische Toxizität sehr gering ist und ihre klinische Effektivität sehr gut an Elektroytveränderungen im Plasma, zum Beispiel im Rahmen eines Cross-over-Protokolls, erfasst werden kann (17).
Auch wenn Schwierigkeiten im Rahmen von Bioäquivalenz-Prüfungen und Aut-idem-Umstellungen bestehen (27): Die klinische Bedeutung von NBCD bei gleichzeitiger Abgrenzung zu Generika beziehungsweise Biosimilars darf nicht unterschätzt werden (27), wobei die parenteral zu applizierenden Eisen-III-Kohlenhydratkomplexe (1), liposomale Formulierungen (2) und Glatiramer(oide) (3) hier hervorzuheben sind.
Dezentrale Zulassungsverfahren von NBCD-Produkten eines Zweitanbieters (»NBCD-Follow-up-Produkte«) auf der Basis physikalisch-chemischer Charakterisierungen ohne weitergehende klinische Prüfungen sind nicht zielführend, wenn man hohe Ansprüche an eine vergleichbare klinische Wirksamkeit, Verträglichkeit und Sicherheit stellt. Damit sind regulatorisch Hybridzulassungen für parenteral zu applizierende Kategorie-1-NBCD anzustreben.
Hans-Peter Lipp studierte von 1983 bis 1987 Pharmazie an der Universität Tübingen. Nach dem dritten Staatsexamen und der Approbation 1988 sowie der Promotion am Institut für Toxikologie (Leitung: Prof. Dr. K. W. Bock) zum Thema »Toxizitätsbeurteilung von komplexen Dioxingemischen« 1991 war er ein weiteres Jahr als Wissenschaftlicher Assistent am Institut für Toxikologie, sodann, von 1992 bis 1998, als Krankenhausapotheker an der Universitätsklinik Tübingen tätig. 1997 wurde er zum Fachapotheker für klinische Pharmazie ernannt. Seit 1998 ist Lipp Chefapotheker des Universitätsklinikums Tübingen, seit 2015 Honorarprofessor der Universität Tübingen. Lipp ist Herausgeber, Mitherausgeber, Autor und Co-Autor von zahlreichen wissenschaftlichen Publikationen und Fachbüchern zur Klinischen Onkologie, Hämostaseologie und Infektiologie. Seine wissenschaftlichen Arbeiten fanden Anerkennung durch viele Preise und Ehrungen.


