 |  | Hans-Peter Lipp |
 |
09.05.2021 08:00 Uhr |
Bis heute ist es schwierig, nicht zuletzt mit Blick auf Fragen der Zulassung, eine einfache Definition für alle NBCD-Vertreter zu finden. / Foto: Adobe Stock/nmann77
Tatsächlich ist es bis heute schwierig, nicht zuletzt auch mit Blick auf Fragen der Zulassung, eine einfache Definition für alle NBCD-Vertreter zu finden, da die betroffenen Arzneimittel keine in sich homogene Gruppe darstellen. Mehrere Experten haben deshalb in der Vergangenheit vielfach versucht, einzelne NBCD in weitere Untergruppen zu kategorisieren (Tabelle 1), um sie systematisch gegenüber anderen Vertretern besser abgrenzen zu können (1–5).
| Kategorie | INN-Bezeichnungen (Auswahl) | Referenzarzneimittel |
|---|---|---|
| 1Nanotechnologisch hergestellte Wirkstoffkomplexe mit schwer zu charakterisierenden oligo- und polymeren Wirkstoffen | Glatirameracetat (s.c.) | Copaxone® |
| 1Nanotechnologisch hergestellte Wirkstoffkomplexe mit schwer zu charakterisierenden oligo- und polymeren Wirkstoffen | Eisen(III)-Sucrose (i.v.) et cetera | Venofer® |
| 1Nanotechnologisch hergestellte Wirkstoffkomplexe mit schwer zu charakterisierenden oligo- und polymeren Wirkstoffen | Sevelamer p.o. | Renvela® |
| 2komplexe liposomale Einbettungen mit genau definierten Wirkstoffen und Albuminkonjugate | Liposomales Amphotericin B (i.v.) | AmBisome® |
| 2komplexe liposomale Einbettungen mit genau definierten Wirkstoffen und Albuminkonjugate | Liposomal pegyliertes Doxorubicin (i.v.) | Caelyx® |
| 2komplexe liposomale Einbettungen mit genau definierten Wirkstoffen und Albuminkonjugate | Liposomales Cytarabin (i.th.) | DepoCyte® (a.H.) |
| 2komplexe liposomale Einbettungen mit genau definierten Wirkstoffen und Albuminkonjugate | Liposomales Irinotecan (i.v.) | Onivyde® |
| 2komplexe liposomale Einbettungen mit genau definierten Wirkstoffen und Albuminkonjugate | Nab-Paclitaxel (i.v.) | Abraxane® |
| ≥3(komplexe emulgierte Einbettungen genau definierter Wirkstoffe zur lokalen Applikation) | Ciclosporin-Emulsion (AT) | Restasis® ATIkervis® |
| ≥3komplexe emulgierte Einbettungen genau definierter Wirkstoffe zur lokalen Applikation | mRNA-basierte Impfstoffe (eingebettet in Lipid-Nanopartikel) | z. B. Comirnaty® |
So finden sich in der Kategorie 1 der derzeit diskutierten NBCD-Klassifikation (1) das Oligopeptidgemisch Glatiramer und die intravenös applizierbaren Eisen-III-Kohlenhydratkomplexe (2) sowie das Allylamin-Polymer Sevelamer (1).
Wird »Glatirameracetat« seit mehr als 20 Jahren in der Behandlung der Multiplen Sklerose (MS) als subkutane Therapie eingesetzt, so handelt es sich um ein Gemisch (Acetatsalz) aus synthetischen Polypeptiden mit L-Glutamin, L-Lysin, L-Alanin und L-Tyrosin und einem Streubereich des Molekulargewichts zwischen 4,7 und 13 kDa.

Oftmals werden Zweitanbieter aufgefordert, Ergebnisse aus Vergleichsstudien vorzulegen, die die Nicht-Unterlegenheit des NBCD gegenüber dem Referenzarzneimittel belegen. / Foto: Adobe Stock/BillionPhotos.com
Mit Glatiramer wird also eine Mischung immunogen wirksamer Polypeptide variabler Sequenz und Größe beschrieben, deren exakte Charakterisierung mit der verfügbaren State-of-the-art-Analytik nicht möglich ist. Aus diesem Grund kann nur über einen streng festgelegten Herstellungsprozess die Antigen-Homologie mit daraus resultierender klinischer Wirksamkeit und Sicherheit von Charge zu Charge gewährleistet werden (6).
Als im Jahr 2016 das Nachfolgeprodukt Clift® neben dem Referenzarzneimittel Copaxone® auf dem EU-Markt erschien, war zunächst eine gewisse Unsicherheit unter MS-Patienten hinsichtlich der Frage zu erkennen, ob die Anwendung eines Produkts eines Zweitanbieters ausreichend effektiv, verträglich und sicher sei.
Angesichts der Komplexität des Produkts war gemäß Artikel 10 (3) der Direktive 2001/83/EC allerdings im Vorfeld festgelegt worden, dass eine Zulassung von Glatiramer wie bei den Generika mit Bioäquivalenz-Prüfung an gesunden Probanden nicht zielführend ist, sodass die Vorgaben für eine Hybridzulassung anzuwenden seien.
In diesem Zusammenhang war der Zweitanbieter aufgefordert worden, die Vorgaben, wie sie bei einer Biosimilar-Zulassung zur Anwendung kommen, zu berücksichtigen. Somit musste der Zweitanbieter – im Gegensatz zu einer Generikazulassung – auch Ergebnisse einer direkten doppelblinden Phase-III-Vergleichsstudie mit dem Referenzarzneimittel Copaxone® bei MS-Patienten für die Zulassung vorlegen, die die Nicht-Unterlegenheit des NBCD über den Beobachtungszeitraum von neun Monaten belegten.
Darüber hinaus erfolgte nach Ablauf dieser Zeitspanne noch eine offene Nachbeobachtungsphase über weitere 15 Monate. Diese GATE-Studie hatte zum Ergebnis, dass das Glatiramer-haltige Arzneimittel des Zweitanbieters dem Referenzarzneimittel Copaxone® in den Endpunkten klinische Wirksamkeit, Verträglichkeit und Sicherheit nicht unterlegen ist (7, 8).

Auch wenn zunächst eine direkte Austauschbarkeit möglich zu sein scheint, können weiterführende Analysen durchaus noch auf Produktdifferenzen verweisen. / Foto: Adobe Stock/yanlev
Auch wenn damit zunächst eine direkte Austauschbarkeit möglich erscheint, verweist eine Expertengruppe auf weitergehende Ergebnisse ihrer hochauflösenden Peptidanalytik, in der sie auf durchaus noch bestehende Produktdifferenzen zwischen den Chargen des Produkts des Zweitanbieters und dem Referenzarzneimittel hinweist. Diese können aus ihrer Sicht immunologisch relevante Unterschiede unter anderem hinsichtlich der STAT-5-Signaltransduktion via Interleukin-2- beziehungsweise TNFα-Stimulation mit sich bringen (7, 9).
Die endgültige Bewertung einer möglichen klinischen Relevanz dieser In-vitro-Befunde würde allerdings eine sehr hohe Patientenzahl mit einer deutlich längeren Behandlungsdauer notwendig werden lassen. Aus diesem Grund ist es nachvollziehbar, wenn Experten hier von einem »Nanosimilar« anstelle von einem »NBCD-Generikum« sprechen, da eine 100-prozentige Übereinstimmung der komplexen Produkte praktisch nicht erreicht werden kann (1–5).
Sehr viele Diskussionen wurden in den letzten Jahren auch über die intravenös applizierbaren Eisen-III-Kohlenhydratkomplexe als NBCD beziehungsweise Nanopharmazeutika geführt (9, 10). Mittlerweile stehen verschiedene Generationen entsprechender Komplexe zur Verfügung, die sich vor allem in ihren physiko-chemischen, pharmakologischen und klinischen Eigenschaften unterscheiden (Tabelle 2).
| Eisen(III)-Carboxymaltose | Eisen(III)-Derisomaltose | Ferumoxytol | Eisen(III)-Sucrose | Eisen(III)-Dextran | Eisen(III)-Gluconat | |
|---|---|---|---|---|---|---|
| Handelsname | Ferinject®(Injectafer®) | Monofer®(Diafer®) | (Feraheme®)(Rienso®) | Venofer®Fermed® (ISS) | Cosmofer®(INFeD®) | Ferrlecit® |
| Verfügbare Ampullengröße | 100, 500, 1000 mg | 100, 500, 1000 mg | 100 mg | 100 mg | 40, 62,5 mg | |
| max. ED i.v. | 1000 mg | 20 mg/kg | 510 mg | 200 mg** | 20 mg/kg | 62,5 (-125) mg |
| Infusionsdauer | ≥15 min | ≥15 min | 15 min | 30 min** | 60 min-6 h | 10-60 min |
| Molekulargewicht (kDa) | 150 | 69 | 185 | 43,3 | 103 | 37,5 |
| Relative Komplexstabilität | hoch | hoch | hoch | mittel | hoch | niedrig |
| Dialysierbares Eisen (%) | <0,002 | <0,002 | <0,002 | 0,057 | 0,1 | 0,789 |
| Dextrangehalt bzw. dextranbasiert | nein | (nein)* | ja | nein | ja | nein |
| Plasma-HWZ [h] | 7-12 | 20 | circa 15 | 6 | 5-20 | circa 1 |
* Dextrane sind als sonstige Bestandteile nicht aufgeführt. Der Nachweis einer Antidextran-Reaktivität mittels Immunodiffusionsassay wird unter Experten weiterhin kontrovers diskutiert.
** Wird die Infusionszeit auf mindestens 3,5 Stunden verlängert, beträgt die Tageshöchstdosis bei maximal wöchentlicher Anwendung 500 mg. Die Angaben der Handelsnamen in Klammern beziehen sich auf Fertigarzneimittel, die außerhalb der EU zugelassen sind.
Dabei spielt zum einen die feste Einbettung des Eisenoxidhydroxid-Kerns im Zentrum des ihn umgebenden Kohlenhydratgerüsts eine zentrale Rolle. Zum anderen ist aber auch die Wahl der Kohlenhydratzusammensetzung selbst von klinischer Relevanz, da sich hieraus unterschiedliche Inzidenzen an Überempfindlichkeitsreaktionen (Hypersensitivitätsreaktion = HSR) beziehungsweise an Hypophosphatämien nach intravenöser Eisengabe ergeben können (11–14).
Dass diese Komplexe, also zum Beispiel Fe(III)-Carboxymaltose versus Fe(III)-Derisomaltose, nicht einfach 1:1 gegeneinander austauschbar sind, ist nicht nur durch die unterschiedlichen Bezeichnungen der Internationalen Freinamen (INN), sondern auch durch die verschiedenen Studienergebnisse im Cross-over-Design nachvollziehbar. So war produktabhängig, zum Beispiel im Vergleich von Fe(III)-Carboxymaltose versus Fe(III)-Derisomaltose, eine unterschiedliche Häufigkeit an HSR und Hypophosphatämien nach intravenöser Gabe beobachtet worden (11–13).
Allerdings zeigten auch Studien mit intravenös zu applizierenden Fe(III)-Sucrose-Präparaten gleicher INN Abweichungen in der klinischen Wirksamkeit und Verträglichkeit zwischen dem Produkt eines Zweitanbieters und dem Referenzarzneimittel (Venofer®). Diese Beobachtungen erlauben die Hypothese, dass trotz der identischen chemischen Zusammensetzung unterschiedliche klinische Effekte erzielt werden können, die letztendlich auf die strukturellen Eigenschaften der Nanopartikel und deren Herstellungsprozess zurückzuführen sind und den Begriff der Iron-Sucrose-Similars (ISS) in der Fachliteratur mit sich gebracht haben. Die geäußerte Kritik an den nationalen Zulassungsverfahren für ISS ist deshalb nachvollziehbar (15, 16).
Die klinische Effektivität der Produkte lässt sich bisher nur mit der Steigerung von Hämoglobinwerten (ΔHb) oder der prozentualen Transferrin-Sättigung (ΔTSAT%) über die Zeit als Surrogatparameter erfassen, was aufgrund der inter- und intraindividuellen Schwankungsbreite dieses Surrogatparameters einer präzisen direkten Vergleichbarkeit dieser Produkte Grenzen setzt.
Eine Bioäquivalenzprüfung, das heißt pharmakokinetische Prüfung bei Bestimmung von cmax und AUC parenteraler Eisen-III-Kohlenhydratkomplexe ist wiederum nicht möglich (10). Mögliche signifikante Unterschiede in der Häufigkeit klinisch relevanter, schwerer HSR sind wiederum nur mithilfe von Megastudien bei den Dritt-Generations-Komplexen herauszuarbeiten, da entsprechende Inzidenzen bisher zwischen 0,1 bis 0,3 Prozent angegeben werden (14).
NBCD (Non-Biological-Complex-Drugs) sind synthetisch hergestellte Wirkstoffe, die aufgrund ihrer komplexen Struktur nicht vollständig physiko-chemisch charakterisierbar sind. Die pharmakologischen Eigenschaften (Pharmakokinetik und -dynamik) sind vom Herstellungsprozess und der daraus resultierenden Formulierung abhängig. Zu den parenteral applizierbaren NBCD zählen unter anderem liposomal eingebettete Arzneistoffe, Eisen-III-Kohlenhydratkomplexe und Glatiramer.
Nanopharmazeutika sind synthetisch hergestellte Wirkstoffkomplexe, die sich in ihrer Größe im nanomolaren Bereich zwischen 1 bis 100 nm bewegen, wobei die Nanodimension zur pharmakologischen Aktivität beiträgt. Aufgrund der Komplexität dieser Nanopartikel und der Schwierigkeit, sie vollständig physiko-chemisch beschreiben zu können, werden sie in der Regel als Subgruppe der NBCD klassifiziert.
In der Klasse-1-Kategorisierung von NBCD finden sich auch die Wirkstoffe Sevelamercarbonat beziehungsweise Sevelamerhydrochlorid (Renvela® beziehungsweise Renagel®). Dabei handelt es sich um Polymere von Allylaminen, die mittels Epichlorhydrin quervernetzt wurden. Die Wirkstoffe werden bei Hyperphosphatämien als Phosphatbinder oral eingesetzt, um den Phosphateintrag über die Nahrung deutlich zu senken.
Auf Gramm-Basis und in der Dosierung besteht kein Unterschied zwischen Sevelamercarbonat und -hydrochlorid, jedoch weist Ersteres eine höhere Pufferkapazität auf. Es ist davon auszugehen, dass aufgrund der hochmolekularen komplexen Struktur des Polymers keine nennenswerte Wirkstoffabsorption nach oraler Gabe erfolgt (17).
Sevelamer unterscheidet sich von den bisher aufgeführten Klasse-1-NBCD-Produkten dadurch, dass es zum einen nicht parenteral eingesetzt wird sowie zum anderen keine relevante systemische Bioverfügbarkeit nach oraler Gabe aufweist. Es handelt sich allerdings um ein NBCD, wenn man den komplexen Herstellungsprozess und die damit verbundene Schwierigkeit der präzisen Analytik der entstandenen quervernetzten Komplexe als Definition heranzieht (17).

Zu beachten sind auch die den NBCD zugrunde liegenden komplexen Herstellungsprozesse. / Foto: Adobe Stock/Ivan Traimak
Sowohl im niedergelassenen Bereich als auch in Krankenhäusern erfolgte nach Einführung des Sevelamer-Nanosimilars von Zweitanbietern (zum Beispiel Sevemed®) eine relativ rasche, vor allem Preis-getriggerte Umstellung. Eine Diskussion wie bei den parenteral anzuwendenden Eisen-III-Kohlenhydratkomplexen oder Glatiramer fand nicht statt.
Allerdings ist dabei zu berücksichtigen, dass ein nicht absorbierbarer Komplex im Rahmen der Arzneimitteltherapiesicherheit (AMTS) anders einzuordnen ist, zumal die engmaschige Bestimmung der Phosphatspiegel im Serum bei entsprechenden Umstellungen einen relativ verlässlichen Anhaltspunkt bietet, wie die klinische Wirksamkeit des Präparats im Rahmen einer Umstellung zu bewerten ist.
In der Kategorie 2 der NBCD-Klassifikation finden sich vor allem parenteral applizierbare liposomale Formulierungen wie zum Beispiel das Polyenantibiotikum Amphotericin B oder die Zytostatika Daunorubicin, Cytarabin, Doxorubicin, Irinotecan und Vincristin (1, 18).
Wie schwierig sich liposomale Einbettungen im Herstellungsprozess gestalten können und mit welchen beträchtlichen Schwierigkeiten im Rahmen der Erzielung einer möglichst geringen Chargenvariabilität, homogenen Inkorporation und exakten Produktanalytik zu rechnen ist, wurde insbesondere am Fertigarzneimittel DepoCyte® deutlich (19).
Während der analytische Nachweis des Antimetaboliten Cytarabin vergleichsweise unproblematisch ist, kommt es bei DepoCyte® vor allem auf die allmähliche Wirkstofffreisetzung aus den multilamellaren Strukturen an. Denn nur so bringt das weiterentwickelte Cytarabin-haltige Fertigarzneimittel den Vorteil mit sich, lediglich alle zwei Wochen intrathekal mit jeweils 50 mg Cytarabin verabreicht werden zu müssen, während die konventionelle, nicht liposomale Form eine intrathekale Injektion von 40 mg zweimal pro Woche erfordert (18). Damit entwickelte sich DepoCyte® zum Standard in der Behandlung der Meningeosis neoplastica, bevor nicht näher umschriebene »Produktprobleme« zu einem bis heute andauernden kompletten Lieferabriss führten.
Der Fall DepoCyte® macht deutlich, dass die Herstellung von Arzneimitteln mit parenteral applizierbaren Liposomen einer äußerst anspruchsvollen Galenik bedarf und der Nachweis der Freisetzungskinetik der inkorporierten Wirkstoffe sehr hohe Ansprüche an die eingesetzte Analytik stellt. Die Zuordnung zu den NBCD, vor allem wenn sie parenteral verabreicht werden, ist nachvollziehbar (20).
Zwar lassen sich auch topisch applizierbare Liposomen oder Nanoemulsionen mit Wirkstoffen, die zur Anwendung am Auge bestimmt sind, der Gruppe der NBCD zuordnen. Doch können an sie nicht dieselben hohen Anforderungen bei einer Zulassung von Zweitanbietern gestellt werden wie bei parenteralen Formulierungen. Daher werden zum Beispiel entsprechende Ciclosporin-haltige Formulierungen der Gruppe 3 zugeordnet (21).
Auch fortentwickelte Depotformulierungen, zum Beispiel Paliperidonpalmitat zur Erhaltungstherapie bei Schizophrenie, lassen sich prinzipiell den NBCD zuordnen. Allerdings ist in diesem Zusammenhang die Wirkstoffanalytik deutlich besser umsetzbar als bei den genannten Produkten der Klasse 1 und 2, dieses insbesondere wenn Bioäquivalenz-Prüfungen durchgeführt werden müssen.
Dennoch geben Experten immer wieder zu bedenken, dass auch bei inhalativ anzuwendenden Formulierungen wie zum Beispiel Pulverinhalaten, Depotarzneimitteln zur intramuskulären Injektion, Transdermalen Therapeutischen Systemen (TTS) oder Dermatika mit komplexer Galenik im Rahmen einer Aut-idem-Substitution die Besonderheiten der zugrunde gelegten Galenik oft unterschätzt werden (1–5).

Auch bei inhalativ anzuwendenden Formulierungen wie bei Depotarzneimitteln zur intramuskulären Injektion, Transdermalen Therapeutischen Systemen (TTS) oder spezifischen Dermatika werden die Besonderheiten infolge der zugrunde liegenden Galenik häufig unterschätzt. / Foto: Adobe Stock/P&G
Auch die seit Kurzem verfügbaren Covid-19-mRNA-Impfstoffe Comirnaty® und Moderna® sind in Lipid-Nanopartikel eingebettet und erfüllen damit in gewisser Weise die Voraussetzungen eines NBCD, da die mRNA-Moleküle nicht rekombinant über cDNA-veränderte Zellkulturen, sondern über eine zellfreie In-vitro-Transkription aus entsprechenden DNA-Vorlagen hergestellt werden.
So entsteht nach dem Auftauen und Verdünnen von zum Beispiel Comirnaty® eine weiße bis grauweiße, opake Dispersion, die nicht heftig geschüttelt werden darf, da ansonsten mit einem Austritt der inkorporierten mRNA zu rechnen ist (22). Allerdings stellt sich derzeit bei diesem Produkt patentrechtlich nicht die Frage nach Zweitanbietern.
Verliert ein Wirkstoff innerhalb der EU seinen Patentschutz, so ergibt sich bei den meisten Substanzen zunächst die Frage, ob es sich um eine chemisch genau definierbare Einzelsubstanz oder um ein rekombinant gewonnenes (Glyko)protein (Biological) handelt.
Im ersten Fall befindet man sich auf regulatorischer Ebene im Bereich der Generika, im letzteren Fall im Bereich der Biosimilars, deren Zulassung innerhalb der EU ausschließlich zentral über die European Medicines Agency (EMA) erfolgen kann (Grafik). Eine Sonderstellung nehmen in diesem Zusammenhang die niedermolekularen Heparine (NMH) ein, da sie regulatorisch den Biosimilars zugeordnet werden, selbst aber keine Glykoproteine, sondern partialsynthetisch gewonnene Folgeprodukte des unfraktionierten Heparins darstellen und damit biologischen Ursprungs sind. Im Gegensatz zu den rekombinanten Produkten erfordert die Zulassung eines NMH-Biosimilars allerdings nicht zwingend eine Phase-III-Studie im direkten Vergleich mit einem Referenzarzneimittel in einem klinisch relevanten Anwendungsgebiet (23).
Auch wenn mit dieser Kategorisierung die Zulassung eines Großteils von Arzneimitteln nach Patenschutzverlust klar geregelt ist, ergeben sich insbesondere für die Stoffklasse der NBCD noch eine Reihe offener Fragen, da ihre substanzspezifischen Charakteristika im Rahmen der genannten etablierten Zulassungsprozesse für Generika bisher nicht ausreichend berücksichtigt werden.
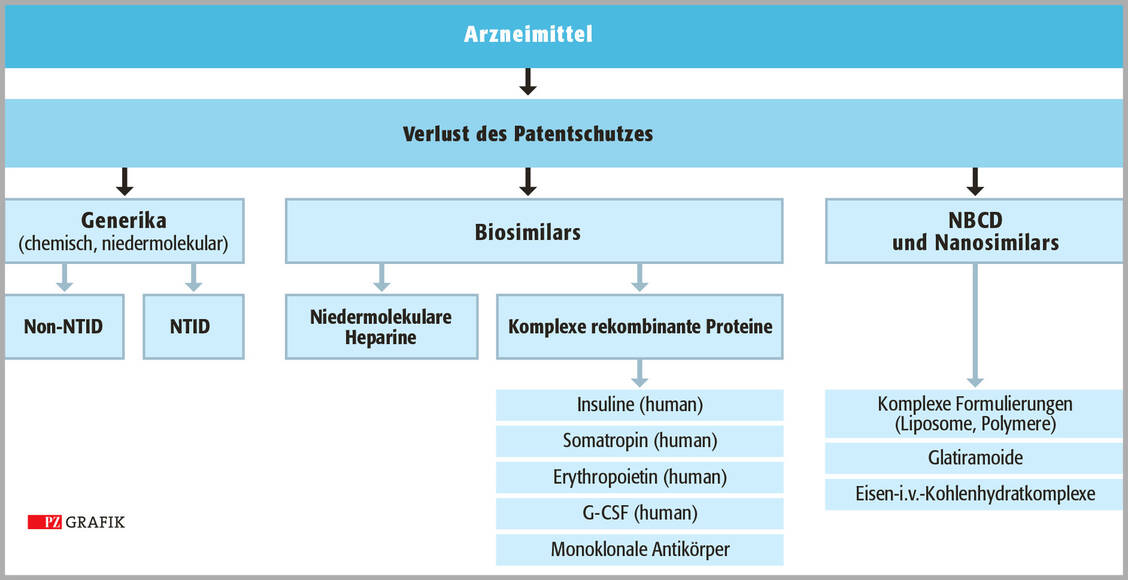
Die derzeit diskutierte Einteilung und Unterscheidung von Arzneimitteln nach Patentschutzverlust in Generika, Biosimilars und NBCD beziehungsweise NBCD-Similars. NTID = Generika mit geringer therapeutischer Breite (mod. nach 28) / Foto: PZ/Stephan Spitzer
Wird in diesem Zusammenhang häufig auch der Begriff Nanopharmazeutika zur weitergehenden Produktbeschreibung der NBCD verwendet, so weisen ihre hinsichtlich der pharmakologischen Aktivität bedeutsamen Nanopartikel in der Summe ihrer inneren und äußeren Dimension eine Größe zwischen 1 nm und 100 nm auf und liegen teilweise in einem wässrigen Medium in kolloidal gelöster Form vor. Dabei trägt die Nanodimension zu den pharmakologischen Effekten des NBCD bei (1–5).
Wird ein Fertigarzneimittel innerhalb der EU zugelassen, so entscheidet zunächst die Art des Wirkstoffs, inwieweit eine zentrale Zulassung über die EMA erfolgen muss. Hierzu zählen sämtliche rekombinant hergestellten Arzneimittel, alle Gen- beziehungsweise Zelltherapeutika, Orphan Drugs, Virustatika und Humanarzneimittel, die einen innovativen Ansatz zur Behandlung von Krebserkrankungen, Diabetes mellitus, neurodegenerativen oder autoimmun-bedingten Erkrankungen bieten. Für die rekombinant gewonnenen Wirkstoffe spielt es damit keine Rolle, ob es sich um Produkte eines Erst- oder Zweitanbieters (Biosimilar) handelt.
»The process makes the product«: Zwar gelten auch für NBCD die Zulassungsbedingungen ähnlich denen der Biologicals und Biosimilars. Die Zulassung für das Produkt eines Zweitanbieters kann aber bisher dezentral auf rein nationaler Ebene erfolgen, da die NBCD per se bisher nicht Gegenstand eines verpflichtend zentralen Zulassungsprozesses sind, es sei denn, die Art des Wirkstoffs macht dies erforderlich (1–5, 24).

Verliert ein Wirkstoff innerhalb der EU seinen Patentschutz, so ergibt sich bei den meisten Substanzen zunächst die Fragestellung, ob es sich um eine chemisch genau definierbare Einzelsubstanz oder um ein rekombinant gewonnenes (Glyko)protein handelt. / Foto: Adobe Stock/Grecaud Paul
Um es noch einmal zusammenzufassen: Das NBCD Glatiramer ist als Ergebnis eines komplexen synthetischen Herstellungsverfahrens zu verstehen, wobei die einzelnen Herstellungsschritte am Ende entscheidenden Einfluss auf die genaue Zusammensetzung des Endprodukts haben.
Eine Bioäquivalenz-Prüfung wie bei einer Generika-Zulassung ist nicht durchführbar, da die einzelnen Polypeptide im Plasma nicht erfasst werden können. Der Nachweis einer Immunmodulation auf der Basis von Surrogatparametern bei gesunden Probanden hätte wiederum eine zu ungenaue Aussagekraft für die Anwendung bei Multipler Sklerose (MS) zur Folge gehabt.
Damit wurde letztendlich der Weg für eine Hybridzulassung geebnet; in diesem Fall wurde – im Gegensatz zu einer Generika-Zulassung – eine Phase-III-Vergleichsstudie gegenüber dem Referenzarzneimittel Copaxone® für die Zulassung des Nachfolgepräparats (zum Beispiel Clift®) von der EMA verpflichtend vorgeschrieben, ähnlich wie bei den Biosimilars (7–9, 25).
Der zentrale Eisen-III-Oxid-Hydroxid-Kern bei Eisen-III-Kohlenhydratkomplexen ist bei den Präparaten der dritten Generation fest in die jeweiligen Kohlenhydratstrukturen eingebettet, sodass am Ende eine hochkomplexe nanokolloidale Struktur entsteht, die über konventionelle analytische Methoden nicht mehr ausreichend genau präparatespezifisch charakterisiert werden kann. Hinzu kommt eine gewisse Mikroheterogenität des Endprodukts von Charge zu Charge – ähnlich wie bei den Biologicals und Biosimilars (1–5).
Bisher können Produkte von Zweitanbietern, zum Beispiel Fe(III)-Sucrose, im Gegensatz zu Biosimilars national zugelassen werden. Da eine Bioäquivalenz-Prüfung im Rahmen der verwendeten Komplexe im Plasma analytisch nicht möglich ist, müssen zwangsläufig Surrogatparameter wie zum Beispiel ∆TSAT% oder ∆Hb für vergleichende Untersuchungen herangezogen werden. Erfahrungen aus der Praxis haben allerdings gezeigt, dass im Rahmen eines Aut-idem-Wechsels von einem Referenzarzneimittel (Venofer®) auf ein ISS Probleme in der Verträglichkeit und Wirksamkeit auftraten (26).
Experten fordern deshalb seit Längerem, dass nach einem Patenschutzverlust von parenteralen Eisen-III-Kohlenhydratkomplexen Produkte von Zweitanbietern ebenfalls eine Hybridzulassung – ähnlich wie bei Glatiramer – durchlaufen müssen, um die klinische Nicht-Unterlegenheit des Nanopharmazeutikums gegenüber dem Referenzarzneimittel fundierter zu belegen.
Diese Forderung ist auch im Einklang mit dem »Reflection Paper« des »Committee for Medicinal Products for Human Use« (CHMP) der EMA, das physikalisch-chemische Produktcharakterisierungen als Nachweis für eine möglichst hohe Übereinstimmung mit bereits zugelassenen Präparaten unter evidenzbasierten Aspekten als nicht ausreichend betrachtet (1–5).
Ob damit ein substanzspezifisch unterschiedliches Risiko für schwere HSR Grad ≥ 3 tatsächlich herausgearbeitet werden kann, bleibt offen, da sicher eine relativ hohe Zahl an Patienten notwendig sein dürfte, um eine signifikante Differenz zwischen zwei Behandlungsgruppen herausarbeiten zu können.
Hingegen lassen sich unterschiedliche Inzidenzen an Grad ≥ 1 HSR zweifelsohne relativ gut erfassen (11, 14). Die klinische Bedeutung der HSR darf in diesem Zusammenhang nicht unterschätzt werden, da das Auftreten einer schweren lebensbedrohlichen Form keine weitere Anwendung eines intravenös zu applizierenden Eisen-III-Kohlenhydratkomplexes jedweder Art erlaubt (14).
Sehr komplex sind zweifelsohne auch die parenteral zu applizierenden liposomalen Einbettungen mit Amphotericin B, Anthrazyklinen oder Irinotecan. Obwohl für einige Präparate das Patent schon seit Längerem abgelaufen ist, stehen bis heute keine entsprechenden Nanosimilars, also Produkte von Zweitanbietern, innerhalb der EU zur Verfügung.
Dieses dürfte auch mit ihrer Komplexität und notwendigen Chargenkonformität in Zusammenhang stehen. Da dem frei verfügbaren, prozentualen Wirkstoffanteil eine maßgebliche Bedeutung im Toxizitätsprofil der Formulierung zukommt, sind auch in diesem Fall hohe Ansprüche an das Zulassungsverfahren für Zweitanbieter zu stellen (18).
Hingegen wird man – trotz ebenfalls hoher Wirkstoffkomplexität – nicht dieselben Ansprüche an nicht resorbierbare NBCD wie zum Beispiel Sevelamer stellen, da ihre systemische Toxizität sehr gering ist und ihre klinische Effektivität sehr gut an Elektroytveränderungen im Plasma, zum Beispiel im Rahmen eines Cross-over-Protokolls, erfasst werden kann (17).
Auch wenn Schwierigkeiten im Rahmen von Bioäquivalenz-Prüfungen und Aut-idem-Umstellungen bestehen (27): Die klinische Bedeutung von NBCD bei gleichzeitiger Abgrenzung zu Generika beziehungsweise Biosimilars darf nicht unterschätzt werden (27), wobei die parenteral zu applizierenden Eisen-III-Kohlenhydratkomplexe (1), liposomale Formulierungen (2) und Glatiramer(oide) (3) hier hervorzuheben sind.
Dezentrale Zulassungsverfahren von NBCD-Produkten eines Zweitanbieters (»NBCD-Follow-up-Produkte«) auf der Basis physikalisch-chemischer Charakterisierungen ohne weitergehende klinische Prüfungen sind nicht zielführend, wenn man hohe Ansprüche an eine vergleichbare klinische Wirksamkeit, Verträglichkeit und Sicherheit stellt. Damit sind regulatorisch Hybridzulassungen für parenteral zu applizierende Kategorie-1-NBCD anzustreben.
Hans-Peter Lipp studierte von 1983 bis 1987 Pharmazie an der Universität Tübingen. Nach dem dritten Staatsexamen und der Approbation 1988 sowie der Promotion am Institut für Toxikologie (Leitung: Prof. Dr. K. W. Bock) zum Thema »Toxizitätsbeurteilung von komplexen Dioxingemischen« 1991 war er ein weiteres Jahr als Wissenschaftlicher Assistent am Institut für Toxikologie, sodann, von 1992 bis 1998, als Krankenhausapotheker an der Universitätsklinik Tübingen tätig. 1997 wurde er zum Fachapotheker für klinische Pharmazie ernannt. Seit 1998 ist Lipp Chefapotheker des Universitätsklinikums Tübingen, seit 2015 Honorarprofessor der Universität Tübingen. Lipp ist Herausgeber, Mitherausgeber, Autor und Co-Autor von zahlreichen wissenschaftlichen Publikationen und Fachbüchern zur Klinischen Onkologie, Hämostaseologie und Infektiologie. Seine wissenschaftlichen Arbeiten fanden Anerkennung durch viele Preise und Ehrungen.


