 |  | Daniela Hüttemann |
 |
19.03.2023 08:00 Uhr |

Hoch individuelle Therapie: Bei der Gentherapie werden fehlerhafte DNA-Sequenzen repariert, ersetzt oder entfernt. / Foto: Getty Images/Eugene Mymrin
Definitionsgemäß werden bei einer Gentherapie menschliche Gene, die durch bestimmte Defekte oder Mutationen Krankheiten bedingen, korrigiert. Die fehlerhafte DNA-Sequenz wird mithilfe von rekombinanten Nukleinsäuren reguliert, repariert, ersetzt, hinzugefügt oder entfernt. Dabei kann die therapeutische DNA als ringförmiges Episom im Zellkern neben der körpereigenen DNA separat vorliegen und abgelesen werden (Grafik, linker Teil). Sie wird also nicht ins Genom integriert: Die körpereigene DNA bleibt unberührt. Es kann aber auch die körpereigene DNA modifiziert werden, zum Beispiel durch Ausschneiden der defekten Stelle und Einfügen eines designten DNA-Stücks (Grafik, rechte Seite) oder durch den Einbau als Vektor im Sinn einer Genfähre.
»Wir haben mittlerweile sogar eine Art Radiergummi, um die fehlerhafte Base wegzuradieren und zu ersetzen«, erklärte Professor Dr. Axel Schambach, Direktor des Instituts für experimentelle Hämatologie an der Medizinischen Hochschule Hannover, vergangenen Herbst bei einem Fortbildungskongress der Apothekerkammer Schleswig-Holstein zum Thema Genetik.

Prinzipien der In-vivo- und Ex-vivo-Gentherapie / Foto: PZ/Stephan Spitzer
Nicht unter den Begriff Gentherapie fallen dagegen andere moderne Behandlungsmöglichkeiten wie Sirane, die auf RNA-Interferenz beruhen, oder mRNA-basierte Impfstoffe, denn sie verändern nicht das Erbgut. Dies wird und wurde von Coronagegnern oft falsch dargestellt.
Bei den ersten gentherapeutischen Versuchen sei es relativ häufig zu schweren Nebenwirkungen gekommen, da virale Vektoren Probleme machten oder bestimmte Promoter-Enhancer-Sequenzen zu Fehlregulationen benachbarter Gensequenzen führten, so Schambach, der als internationaler Experte für die Gentherapie gilt. Mittlerweile habe man bessere Promotoren und das Vektorendesign sicherheitsverbessert. Und: »Dank Genome Editing können wir heute sehr zielgerichtet und präzise korrigieren, wie mit einer genauen Schere. Anschließend schließen wir die Lücke mit einer Art molekularem Pflaster mit der korrigierten Sequenz.«
Die erste in der EU zugelassene Gentherapie kam vor elf Jahren auf den Markt: Alipogen Tiparvovec (Glybera®) zur Behandlung des familiären Lipoprotein-Lipase-Mangels (LPLD). Nicht aus Sicherheitsgründen, sondern aufgrund der mangelnden Nachfrage nahm der Hersteller Uniqure Biopharma es allerdings 2017 wieder vom Markt, als die vorläufige, auf fünf Jahre befristete Zulassung auslief.
Das ist nach wie vor schwierig. Die meisten zugelassenen Präparate verwenden einen viralen Vektor, meist Adeno-assoziierte Viren (Parvoviren), Lentiviren und Gamma-Retroviren. Bei In-vivo-Ansätzen wird die DNA mitunter als liposomale Nanopartikel formuliert, die über die Leber aufgenommen werden. Das funktioniert besonders gut, wenn die Leberzellen selbst das Target sind, zum Beispiel bei der Hämophilie, da hier die Gerinnungsfaktoren hergestellt werden, oder der PCSK9-Hemmung bei Hyperlipidämie. Die bislang meisten Präparate adressieren Erkrankungen, die mit den Blutzellen zu tun haben, da diese besser zugänglich sind.
Zum Teil ist es erforderlich, das Gentherapeutikum direkt ins Zielorgan zu bringen; zum Beispiel wird Voretigen Neparvovec (Luxturna®) intraokulär in die Subretina injiziert. Als schwierig gilt es, Gehirnzellen zu adressieren, was Gentherapien für Morbus Parkinson, die Alzheimer-Erkrankung und andere neurologische Erkrankungen schwieriger macht.
Derzeit sei die Gentherapie als relativ schwerwiegender und teurer Eingriff noch auf seltene, schwere monogenetische Erkrankungen beschränkt, erläutert Schambach im Gespräch mit der PZ. Meist sei es die letzte Therapieoption, zum Beispiel bei der schweren kombinierten Immundefizienz (SCID).
Die Patienten haben keinerlei funktionierende Immunabwehr und sind in diesem Punkt vergleichbar mit Aids-Patienten im Endstadium – jeder Infekt kann tödlich sein. Die Erkrankung wird auch »Bubble Boy Disease« genannt, da in den 1970er-Jahren in den USA der Patient David Vetter eigens von der NASA eine Art Weltraumanzug bekam und darin wie in einer keimarmen Blase einen Großteil seines kurzen Lebens verbrachte.

Rylae-Ann Poulin wurde mit der Erbkrankheit AADC geboren, bei der das Enzym Aromatische L-Aminosäure-Decarboxylase fehlt. Die Patienten leiden unter Entwicklungsverzögerungen, Bewegungsstörungen und geringer Muskelspannung und sterben meist sehr jung. Rylae-Ann erhielt 2019 das Gentherapeutikum Upstaza®. Auf dem Bild aus dem Januar 2023 nimmt die mittlerweile Fünfjährige ganz normal am Sportunterricht teil. / Foto: Picture Alliance/Associated Press/Sakchai Lalit
Anfang der 2000er-Jahre wurde dann das Baby Rhys Evans aus Wales als erster SCID-Patient mangels passendem Knochenmarkspender gentherapeutisch behandelt – und konnte danach in den Streichelzoo gehen. »Mittlerweile ist er 22 Jahre alt und kann fast alles machen wie Gleichaltrige«, so Schambach. 2016 wurde mit Strimvelis® eine erste Gentherapie gegen SCID zugelassen. Weitere befinden sich in klinischen Studien. »Die Überlebensrate lag in den neueren Studien deutlich über 90 Prozent – bei Patienten, bei denen therapeutisch sonst nichts mehr möglich war.«
Die Verfahren wurden und werden weiter optimiert, auch in Schambachs Labor, das für zwei internationale Studien mit SCID-Patienten sicherheitsoptimierte Vektoren entwickelt hat. »Bislang haben wir hier eine gute Effizienz, in den meisten Fällen erfreulicherweise ohne gravierende Nebenwirkungen«, so Schambach. Allerdings brauche es langfristige Beobachtungen, um die Sicherheit und Wirksamkeit beurteilen zu können. Das müsse bei jedem einzelnen Präparat erfolgen.
Entwickelt der Patient zum Beispiel nach 20 oder 30 Jahren Krebs? Reicht eine einmalige Behandlung ein Leben lang oder braucht er einen weiteren Therapiezyklus? Ist dies überhaupt möglich oder würde der Vektor des Gentherapeutikums von Antikörpern sofort neutralisiert werden, da das Immunsystem ihn wiedererkennt? »Das hängt sehr vom verwendeten Gentherapievektor ab«, so Schambach. »Auch für immunologisch aktivierende Genfähren haben wir mittlerweile Möglichkeiten, einen Genfähren-Ersatz zu finden.«
Bislang geht man davon aus, dass die einmalige Behandlung in vielen Fällen ausreicht, insbesondere wenn die körpereigene DNA modifiziert wird. Liegt die künstliche DNA als Episom vor, könnte sie theoretisch im Lauf der Jahre durch die Zellteilung ausgedünnt werden. Dann könnte die Wirkung nachlassen. »Dieser Effekt würde in sich nicht teilenden Zellen nicht so stark ausgeprägt sein«, erläutert Schambach. Hier sei trotzdem eine zeitlich lange Genexpression möglich.
Solche Unsicherheiten und die hohen Kosten rechtfertigen den Einsatz bislang nur bei schweren, ansonsten unheilbaren Erkrankungen mit niedriger Lebenserwartung, bei denen die Pathogenese eindeutig geklärt und natürlich genetisch bedingt ist.
»Wir machen bei jedem einzelnen Patienten und für jede Erkrankung eine Risiko-Nutzen-Bewertung, immer im Vergleich zum State of the Art«, versichert Schambach. »Nach wie vor ist bei SCID der Goldstandard die Knochenmarktransplantation, aber wenn kein passender Spender gefunden wird, können wir autologe Stammzellen nun genetisch korrigieren.«
Ein anderes Beispiel ist die Beta-Thalassämie, bei der eine Untereinheit des Hämoglobins fehlgebildet wird. Die Folge kann eine ausgeprägte Anämie sein. Mittels Gentherapie wird der Körper in die Lage versetzt, wieder die fetale Beta-Globin-Untereinheit des Hämoglobins zu bilden.
Zwei Jahre lang war ein entsprechendes Gentherapeutikum (Betibeglogen Autotemcel (Zynteglo® von Bluebird Bio) in der EU zugelassen. Es wurde aus wirtschaftlichen Gründen vom Markt genommen, nachdem sein Vertrieb zuvor aufgrund vorläufiger Sicherheitsbedenken, die sich nicht bestätigten, gestoppt worden war. In den USA ist es weiterhin erhältlich.
Die Benennung von Gentherapien gibt die Weltgesundheitsorganisation vor. Die Freinamen von Gentherapeutika bestehen immer aus zwei Teilen. Der erste gibt Hinweise auf das zu reparierende Gen, der zweite auf den Vektor. Zwei Beispiele: Val-octoco-gen Roxa-parvo-vec (Roctavian®) und Ona-semno-gen Abe-parvo-vec (Zolgensma®).
Wenn nach einer ausführlichen Nutzen-Risiko-Bewertung die Entscheidung gefallen ist, dass der Patient für die Gentherapie infrage kommt und umgekehrt, könne es mitunter innerhalb weniger Wochen losgehen, erklärt Schambach. Voraussetzung ist, dass die Gentherapie bereits in der EU zugelassen ist oder die Behandlung im Rahmen einer genehmigten klinischen Studie erfolgt. Die Genfähre muss unter GMP-Bedingungen produziert werden.
Die Patienten werden gründlich durchgecheckt und Begleiterkrankungen ausgeschlossen. Neben einer eindeutigen genetischen Testung wird auf Antikörper gegen den verwendeten Vektor getestet.
Bei einer Ex-vivo-Gentherapie werden zunächst die körpereigenen Zellen per Apherese entnommen und im GMP-Labor gentechnisch verändert. Dies kann Tage bis Wochen dauern. Dabei finden umfangreiche Qualitätskontrollen statt. Der Patient bekommt etwa drei Tage bis eine Woche vor der Rückinfusion der veränderten Zellen eine chemotherapeutische Konditionierung mit Busulfan oder Fludarabin plus Cyclophosphamid – wie bei einer Knochenmarktransplantation. »So machen wir quasi Platz für die neuen Zellen«, erklärt der Experte. Die eigentliche Infusion sei relativ einfach.

Drei Monate alt war Ainsley Cardente, als sie 2019 aufgrund einer spinalen Muskelatrophie mit dem Gentherapeutikum Zolgensma behandelt wurde. / Foto: Imago/ZUMA Wire
Ein In-vivo-Beispiel ist die Therapie mit Onasemnogen Abeparvovec (Zolgensma®) für Kinder mit spinaler Muskelatrophie. Sie bekommen einen Tag vor der Infusion des Gentherapeutikums zunächst eine Dosis Prednisolon oral (1 mg/kg Körpergewicht), da es zu einer Immunreaktion gegen den Vektor kommt, was zu Leberversagen führen kann. Die eigentliche Gentherapie dauert nur 60 Minuten. Die immunmodulatorische Therapie wird über mindestens 30 Tage weitergeführt und unter Kontrolle der Leberwerte schrittweise ausgeschlichen. Dazu gab es erst kürzlich einen Rote-Hand-Brief, der auf die Corticoidtherapie und Leberwert-Kontrolle genauer einging.
»Die Patienten bleiben meist für mehrere Wochen stationär, bis sich alle Werte normalisiert haben, und kommen dann regelmäßig zur Nachsorge, anfangs öfter, später in größeren Abständen«, erklärt Schambach. Vorgeschrieben ist zudem, dass der Therapieerfolg über bis zu 15 Jahre kontrolliert wird.
Beim Management von Gentherapien ist auch die Krankenhausapotheke involviert. »Wenn feststeht, ob und wann ein Patient behandelt wird, werden wir zeitnah informiert«, berichtet Jessica Gunia, Leiterin der Sterilabteilung der MHH-Zentralapotheke, im Gespräch mit der PZ. Beispiel Zolgensma, mit dem Säuglinge und Kleinkinder mit spinaler Muskelatrophie in der Kinderklinik behandelt werden: Zunächst müsse von der Klinik die Kostenübernahme durch die Krankenkasse geklärt werden (diese Gentherapie kostet rund 2 Millionen Euro). Die Dosisberechnung erfolgt nach Körpergewicht durch die behandelnden Ärzte. Diese wird von den Krankenhausapothekern gegengecheckt und anschließend die Bestellung ausgelöst.
»Gentherapeutische Präparate kommen bei uns zwar nicht täglich vor, aber wir haben mittlerweile Routine im Umgang damit entwickelt«, berichtet Gunia. Der Waren- und Dokumentationsprozess sei zwar aufwendiger, die eigentliche Herstellung der Infusion laufe dann aber im Prinzip wie bei anderen aseptisch hergestellten Medikamenten in der Krankenhausapotheke ab. Das Medikament wird in die Reinräume der Sterilherstellung eingeschleust und unter Laminar-Flow-Werkbänken von qualifiziertem pharmazeutischen Personal zubereitet.
Zolgensma wird aus Irland nach Hannover eingeflogen und von einem Spezialkurierdienst direkt an die Klinikapotheke geliefert. Dort wird die spezielle Transportbox mit dem tiefgekühlten Medikament auf Trockeneis nur an bestimmte Personen nach Vorlage des Personalausweises ausgehändigt. »Wir haben dann eine Checkliste, was alles zu überprüfen ist, wie mögliche Schäden an der Transportbox, eine intakte Verpackung, Patienten-ID, Anzahl und passende Befüllung der Vials und die Temperatur-Logdaten«, erläutert Hassan Darkhabani, stellvertretender Leiter der Sterilabteilung.
Der Patient wird in der Regel am Tag vor der geplanten Gabe einbestellt und noch einmal eingehend untersucht. Station und Apotheke telefonieren am nächsten Morgen, ob der Eingriff wirklich stattfinden kann. Dann geht es in die Sterilherstellung. Insgesamt stellt die Behandlung mit Gentherapeutika einen komplexen Prozess dar, in dem Krankenhausapotheke und Klinik interdisziplinär eng zusammenarbeiten.
Laut Schambach laufen bereits mehr als 1500 Studien zum Einsatz von Gentherapeutika weltweit; darunter Proof-of-Concept-Studien bei Retina-Degeneration, Morbus Parkinson und Transplantat-Abstoßungen. Die Hauterkrankung Epidermolysis bullosa konnte bei ersten Patienten bereits topisch mit einer gelbasierten Gentherapie behandelt werden.
Aktuell sind oder waren zehn Präparate in der EU zugelassen (Tabelle). Zu den neuesten Zulassungen Ende 2022 gehören jeweils eine Gentherapie bei Hämophilie A und B. Im EU-Zulassungsverfahren befinden sich Lenadogen Nolparvovec zur Behandlung der seltenen Augenerkrankung Leber’sche Optikus-Atrophie sowie Exagamglogen Autotemcel zur Behandlung der Sichelzellerkrankung (SCD) und der transfusionsabhängigen Beta-Thalassämie (siehe weiter unten). Die FDA geht von 10 bis 20 weiteren gentherapeutischen Zulassungen pro Jahr in den kommenden Jahren aus. »Gentherapeutisch behandelte Patienten werden Ihnen in den Apotheken also immer häufiger begegnen«, so Schambach. Eine Gentherapie könne je nach Art der Erkrankung und Genkorrektur eventuell Auswirkungen auf die Beratung zur herkömmlichen Medikation haben. »Daher halte ich es für wichtig, dass sich Apothekerinnen und Apotheker schon jetzt mit diesen Behandlungsmöglichkeiten auseinandersetzen.«
| Gentherapeutikum INN | Präparat, Hersteller | Indikation | EU-Zulassung |
|---|---|---|---|
| Alipogen Tiparvovec | Glybera®, Uniqure Biopharma | Hyperlipoproteinämie Typ I | 2012, zurückgezogen 2017 |
| Talimogen Laherparepvec | Imlygic®, Amgen | Melanom | 2015 |
| autologe CD34+-angereicherte Zellfraktion | Strimvelis®, Orchard Therapeutics | schwere kombinierte Immundefizienz (SCID) | 2016 |
| Voretigen Neparvovec | Luxturna ®, Spark Therapeutics | Leber’sche kongenitale Amaurose | 2018 |
| Betibeglogen Autotemcel | Zynteglo®, Bluebird Bio | Beta-Thalassämie | 2019, zurückgezogen 2021 |
| Onasemnogen Abeparvovec | Zolgensma®, Novartis | spinale Muskelatrophie | 2020 |
| Atidarsagen Autotemcel | Libmeldy®, Orchard Therapeutics | metachromatische Leukodystrophie | 2020 |
| Elivaldogen Autotemcel | Skysona®, Bluebird Bio | zerebrale Adrenoleukodystrophie | 2021 |
| Eladocagen Exuparvovec | Upstaza®, PTC Therapeutics | AADC-Mangel | 2022 |
| Valoctocogen Roxaparvovec | Roctavian®, Biomarin | Hämophilie A | 2022 |
| Etranacogen Dezaparvovec | Hemgenix®, CSL Behring | Hämophilie B | 2022 |
Als Unterform der Gentherapie lässt sich die CAR-T-Zelltherapie betrachten, bei der die eigenen T-Lymphozyten ex vivo gentechnisch verändert werden. Hier gibt es mittlerweile sechs zugelassene Präparate in der EU, von denen fünf in Deutschland auf dem Markt sind. Alle werden bei verschiedenen Formen von Blutkrebs eingesetzt (lesen Sie dazu auch den Beitrag: CAR-T-Zelltherapie: Hoffnung und Herausforderung).
Bislang kommen Gentherapien nur in spezialisierten Zentren an Unikliniken mit Maximalversorgung zum Einsatz. »Womöglich wird es irgendwann In-vivo-Therapien in Spritzenform für spezialisierte kleinere Zentren und gegebenenfalls auch die ambulante Versorgung geben, die über die öffentlichen Apotheken vertrieben werden«, kann sich Schambach vorstellen.
Wichtig ist, dass keine Keimzellen verändert werden, die auf die nächste Generation übertragen werden. Unseriöses Beispiel ist der chinesische Wissenschaftler Dr. He, der bei einem Zwillingspaar mittels Genschere CRISPR-Cas die T-Helferzellen von Zwillingen im Embryonalstadium so verändert hatte, dass sie resistent gegen HI-Viren sind, was 2018 bekannt wurde und weltweit Kritik ausgelöst hatte.
»Das war gegen alle guten Regeln der wissenschaftlichen Praxis«, unterstreicht Professor Dr. Malte Spielmann, Direktor des Instituts für Humangenetik am Universitätsklinikum Schleswig-Holstein mit Standorten in Kiel und Lübeck, im Gespräch mit der PZ. Forscher He kam ins Gefängnis. Wie es den Kindern heute geht, ist unbekannt.
CRISPR-Cas wird bereits erfolgversprechend zur Korrektur somatischer Zellen eingesetzt. So gelang es einem Team um Professor Dr. Selim Corbacioglu, Leiter der Abteilung für Pädiatrische Hämatologie, Onkologie und Stammzelltransplantation am Uniklinikum Regensburg, das fetale Gamma-Hämoglobin bei Beta-Thalassämie und Sichelzellanämie durch eine einmalige Transfusion ex vivo »gecrisperter« Zellen bei zwei 21-jährigen Patienten so hochzufahren, dass die beiden nicht mehr auf Bluttransfusionen angewiesen sind. Dies berichtete das Team 2021 im »New England Journal of Medicine« (DOI: 10.1056/NEJMoa2031054). Die Firmen Vertex Pharmaceuticals und CRISPR Therapeutics haben Ende Januar 2023 einen Zulassungsantrag in der EU gestellt. Exagamglogen Autotemcel (Exa-cel, CTX001™) wäre die erste zugelassene CRISPR-Cas-Gentherapie.
Zur Behandlung der Transthyretin-Amyloidose (ATTR) wurde CRISPR-Cas bereits beim Menschen und sogar in einem In-vivo-Verfahren eingesetzt (DOI: 10.1056/NEJMoa2107454). Bei dieser Erkrankung kommt es zu Fehlfaltungen des Proteins Transthyretin, vor allem in den Nervenzellen und im Herzen. Hier wurde die mRNA für das Cas9-Protein mit einer einzelsträngigen guide RNA (gRNA), die es an die richtige Stelle im TTR-Gen bringen soll, als Lipidnanopartikel formuliert und infundiert.
»Wir können seltene Erkrankungen mittels CRISPR-Cas bereits effektiv behandeln und das extrem präzise, effektiv und einfach«, fasst Spielmann zusammen. Technisch wäre es mittlerweile sogar möglich, für jeden Patienten mit einem seltenen monogenetischen Defekt eine individuelle Gentherapie zu entwickeln. Aber wie lässt sich das finanzieren? »Das rüttelt an unserem Solidarsystem.«
Die Industrie hat andere Pläne. »Der nächste Schritt werden die Volkskrankheiten sein«, glaubt Spielmann. So will der US-Kardiologe Dr. Sekar Kathiresan, ehemaliger Direktor des Massachusetts General Hospital und Gründer von Verve Therapeutics, kardiovaskuläre Erkrankungen durch Gen-Editierung ähnlich wie bei der ATTR beschrieben heilen. »Dann hätte man einen lebenslangen Schutz vor den Auswirkungen von zu hohem Cholesterol«, ordnet Spielmann den Ansatz ein.
Mit CRISPR-Cas und einer gRNA soll das Enzym PCSK9 (Proprotein-Convertase-Subtilisin/Kexin Typ 9) so verändert werden, dass es weniger LDL-Rezeptoren abbaut und der LDL-Spiegel sinkt – eine einmalige Gentherapie zur dauerhaften Cholesterolsenkung statt lebenslanger PCSK9-Hemmer-Injektionen oder Statin-Einnahme.
In Primaten ist die dauerhafte Lipidsenkung mittels CRISPR-Cas bereits gelungen, wie ein Team um Kathiresan 2021 in »Nature« berichtete (DOI: 10.1038/s41586-021-03534-y). Laut der Website von Verve Therapeutics will man die Therapie nun bei Patienten mit familiärer Hypercholesterolämie und sehr hohen LDL-Werten erproben – das sind laut Firma schätzungsweise 31 Millionen Betroffene weltweit.
Schritt 2 sieht eine Ausweitung auf Patienten ohne diese genetische Vorbelastung, aber mit bestehenden Herz-Kreislauf-Erkrankungen mit hohem Risiko für schwere Ereignisse vor. Im dritten Schritt soll die Therapie bei Erwachsenen mit Risiko für kardiovaskuläre Erkrankungen zum Einsatz kommen – gewissermaßen eine Gentherapie für fast alle und damit ein milliarden-, wenn nicht gar billionenschwerer Markt.

Sollte es künftig massentaugliche Gentherapien geben, wird das Apothekenpersonal immer häufiger damit konfrontiert werden. / Foto: Getty Images/Tom Werner
Die geplante Phase-I-Studie VERVE-101 liege in den USA allerdings auf Eis, da die US-Zulassungsbehörde Bedenken hätte, berichteten US-Medien Anfang Februar. Studienarme in Neuseeland und Großbritannien seien dagegen angelaufen. Erste Daten sollen Mitte des Jahres vorliegen. Der Hannoveraner Arzt und Genforscher Schambach betont: »Auch hier muss eine sorgsame Risiko-Nutzen-Abwägung erfolgen. Die Therapie muss effizient und sicher sein und der Patient über viele Jahre profitieren.«
Nicht mehr weit von einer klinischen Studie entfernt ist ein gentherapeutischer Ansatz bei Diabetes. Affen mit künstlich induziertem Typ-1-Diabetes verhalf eine Gentherapie zu neuen Insulin-produzierenden Betazellen. Die experimentelle Gentherapie GPX-002 der Firma Genprex basiert auf einem Adeno-assoziierten Virus (AVV) mit Alphazellen-spezifischem Promoter, der die Gene pdx1 und MafA per Infusion direkt in die Bauchspeicheldrüse einschleusen soll. Diese Gene kodieren für zwei Transkriptionsfaktoren, die wichtig für die Reifung der Betazellen und die Regulation der Insulinsekretion sind.
Ende Februar 2023 präsentierte die Firma bei einer Diabeteskonferenz in Berlin Daten aus Versuchen mit acht Affen. Der Insulinbedarf der Tiere sank und die Glucosetoleranz verbesserte sich signifikant. Ein Tier erreichte normale Blutzuckerwerte.
Ein anderer Gentherapie-Kandidat mit dem Kürzel GPX-003 soll bei Typ-2-Diabetes die noch vorhandenen Betazellen verjüngen und ergänzen.
Foto: Getty Images/Andrew Brookes
Unter den Begriff »Arzneimittel für neuartige Therapien« (Advanced Therapy Medicinal Products, ATMP) zählen die europäischen Arzneimittelbehörden alle Behandlungsmöglichkeiten, die auf Genen, Geweben oder Zellen basieren. Darunter fallen neben den Gentherapien mit der CAR-T-Zelltherapie als Unterform auch somatische Zelltherapeutika wie Darvadstrocel (Alofisel®, mesenchymale Stromazellen aus dem Fettgewebe von Spendern zur Behandlung von Analfisteln bei Morbus Crohn) und Tabelecleucel (Ebvallo®), das bei Epstein-Barr-positiver Post-Transplantations-proliferativer Erkrankung eingesetzt wird.
Ebenfalls zu den ATMP gehören Gewebeprodukte wie Spherox® und Holoclar®. Spherox enthält sogenannte Sphäroide aus humanen autologen Matrix-assoziierten Chondrozyten und ist zur Reparatur symptomatischer Gelenkknorpeldefekte gedacht. Holoclar enthält lebendes Hornhaut-Gewebeäquivalent und kommt bei Verbrennungen und Verätzungen des Auges in Betracht.
Humangenetiker Spielmann sieht noch ein riesiges Einsatzgebiet für die Gentherapie mit CRISPR-Cas: die Modifikation von tierischen Organen zum Einsatz bei Patienten, die auf ein Spenderorgan warten.
Tatsächlich gab es 2022 den ersten (teil)erfolgreichen Versuch am Menschen. Der 57-jährige David Bennett litt unter einer terminalen Herzerkrankung aufgrund einer viralen Kardiomyopathie. Doch da er früher alkoholkrank war, hatte er keinen Anspruch auf ein Spenderherz.
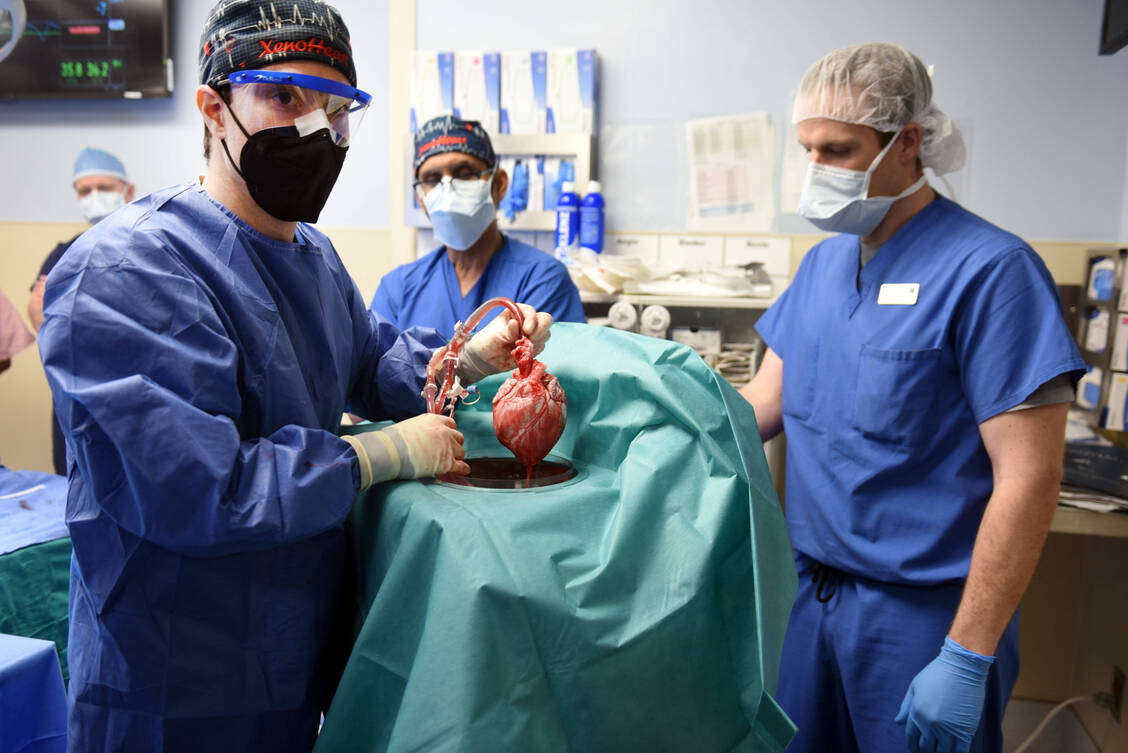
Dieses Schweineherz wurde 2022 mithilfe der CRISPR-Cas-Methode humanisiert und dem Patienten David Bennett eingepflanzt. Bennett überlebte knapp zwei Monate damit. / Foto: Imago/ZUMA Wire
Ein Team um Dr. Bartley Griffith von der University of Maryland ließ ein Schwein von der Firma Revivicor gentechnisch mit zehn CRISPR-Cas-Edits so verändern, dass sein Herz kleiner wuchs und bestimmte Glykosylierungen auf der Oberfläche der Zellen fehlten, die normalerweise das menschliche Immunsystem auf den Plan gerufen hätten (DOI: 10.1056/NEJMoa2201422). Dieses humanisierte Schweineherz wurde dem Patienten im Januar 2022 erfolgreich implantiert. Bennett lebte knapp zwei Monate mit diesem Herzen. 49 Tage nach der Transplantation versagte das Spenderorgan; an Tag 60 wurden lebenserhaltende Maßnahmen abgestellt.
Die Obduktion ergab, dass das Schweineherz ödematös war und sich seine Größe nahezu verdoppelt hatte. Es fanden sich jedoch keine typischen Zeichen einer Transplantat-Abstoßung. Einem Bericht der »New York Times« zufolge wurden bei der Obduktion DNA-Spuren eines Schweinevirus gefunden. Andere Mediziner kommentierten im »New England Journal of Medicine«, dass gerade der Versuch, das Transplantat immunologisch »zu inert« zu machen, paradoxerweise die Abstoßung gefördert haben könnte (»Missing-Self-Phänomen«). Dies ließe sich allerdings mit einer mTOR-Hemmung durch Sirolimus verhindern (DOI: 10.1056/NEJMc2210401).
Griffith und sein Team wollen weitermachen. »Es wäre eine Riesenchance, unser Problem zu lösen, dass wir viel zu wenig Spenderorgane haben«, betont Spielmann. »Daran arbeiten viele Teams und wir werden in zehn Jahren viel weiter sein.«
Daniela Hüttemann studierte Pharmazie an der Philipps-Universität in Marburg. Einen Teil ihres praktischen Jahres forschte sie an der medizinischen Fakultät der National University of Singapore. 2007 erhielt sie die Approbation und begann ein Volontariat bei der Pharmazeutischen Zeitung. Seit 2008 ist sie Redaktionsmitglied und beschäftigt sich besonders mit neuen Themen für die Apothekerschaft, ob pharmazeutische Dienstleistungen, DiGA oder Gentherapien.


