 |  | Sven Siebenand |
 |
12.01.2025 09:00 Uhr |
Eine EU-Zulassungsempfehlung gibt es auch für den Wirkstoff Vilobelimab (Gohibic™) zur Infusion. Gibt die EU-Kommission das finale Go, dann wäre das Medikament zugelassen bei Erwachsenen mit SARS-CoV-2-induziertem akuten Atemnotsyndrom (Acute Respiratory Distress Syndrome, ARDS), die in der Standardbehandlung systemische Corticosteroide erhalten und invasiv mechanisch beatmet werden.
Vilobelimab greift in das Komplementsystem des Körpers ein und ist gegen den Komplementfaktor C5a gerichtet. Schweres Covid-19 geht mit einer starken Komplementaktivierung und der Bildung von C5a einher. Dieses Protein spielt eine Schlüsselrolle bei inflammatorischen Prozessen. Durch die Hemmung von C5a unterbricht der Antikörper die Entzündungskaskade.
Denkbar ist, dass Vilobelimab eines Tages nicht nur beim ARDS im Zuge einer SARS-CoV-2-Infektion zum Einsatz kommt, sondern auch bei ARDS anderer Ursache.
Im Januar erfolgt die Markteinführung der ersten CRISPR/Cas9-basierten Gentherapie: Exagamglogen autotemcel (Exa-cel, Casgevy®) ist zugelassen bei schwerer Sichelzellkrankheit oder transfusionsabhängiger Beta-(β-)Thalassämie.
Die Sichelzellkrankheit ist eine genetische Erkrankung, die zu einer abnormalen Form des Hämoglobins führt, dem sogenannten Sichelhämoglobin. Aufgrund des andersartigen Hämoglobins können die Erythrozyten ihre Elastizität verlieren und kleine Blutgefäße blockieren. Die Betroffenen leiden unter häufigen heftigen Schmerzen, Organschädigungen und einer stark verkürzten Lebenserwartung.
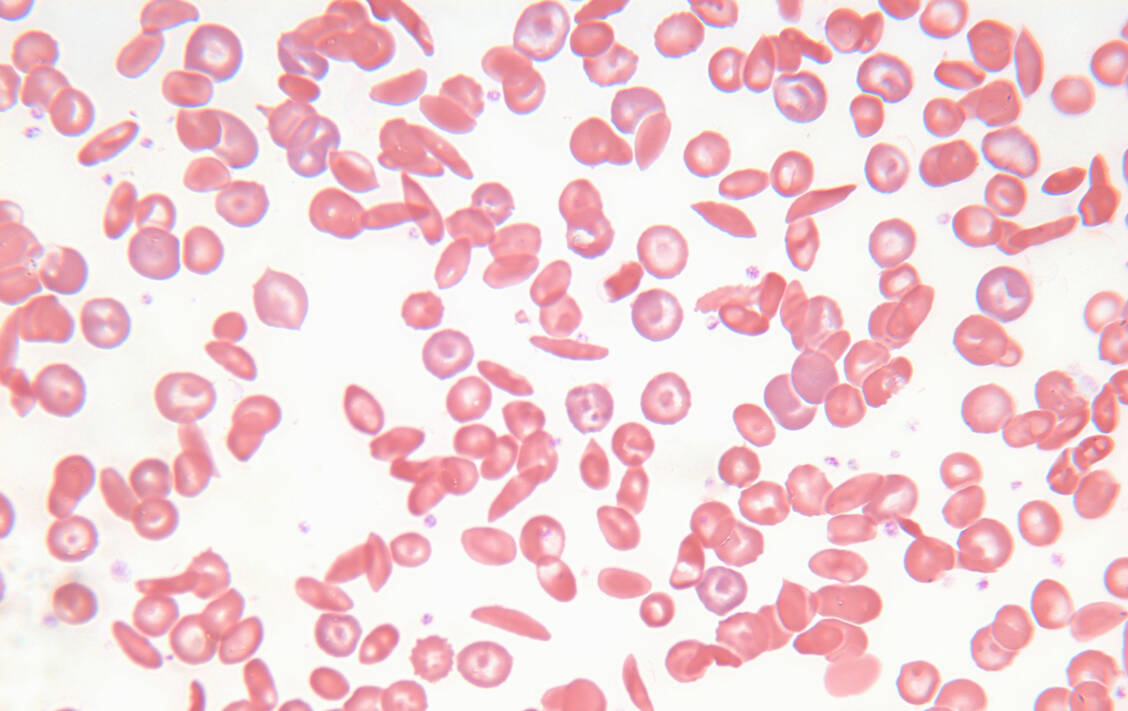
Die Sichelzellkrankheit ist eine genetische Erkrankung, die zu einer abnormalen Form des Hämoglobins führt, dem sogenannten Sichelhämoglobin. Eine CRISPR/Cas9-basierte Gentherapie mit Exagamglogen autotemcel (Exa-cel) könnte die Patienten heilen. / © Getty Images/Connect Images/Michael J. Klein, M.D.
Die β-Thalassämie ist ebenfalls eine genetische Erkrankung. Kennzeichnend ist eine zu geringe oder fehlende Produktion von β-Globin, einem wichtigen Baustein des Hämoglobins; daraus resultiert eine Anämie. Menschen mit schwerer β-Thalassämie sind ihr Leben lang auf Bluttransfusionen angewiesen und haben ebenfalls eine stark reduzierte Lebenserwartung.
Im Rahmen der Gentherapie werden den Patienten die eigenen Stamm- und Vorläuferzellen entnommen und diese ex vivo mit Exa-cel behandelt – die Genschere kommt zum Einsatz. Die sogenannte Erythroid-spezifische Enhancer-Region des BCL11A-Gens wird durch einen präzisen Doppelstrangbruch modifiziert. Das hat zur Folge, dass ein Gen reaktiviert wird, das normalerweise nach der Geburt stillgelegt wird. Werden die modifizierten Zellen dem Patienten zurückinfundiert, produzieren die daraus gebildeten roten Blutkörperchen nun die fetale Hämoglobin-Variante (Hämoglobin F). Sie soll die deformierten Hämoglobin-Varianten ersetzen.


