 |  | Ulrike Holzgrabe |
 |
02.11.2025 08:00 Uhr |
Die derzeitige Behandlung von Schmerzen, insbesondere von chronischen Schmerzen, mit nicht steroidalen Antirheumatika (NSAR) oder Opioiden ist durch schwere Nebenwirkungen sowie Suchtpotenzial geprägt. Hinzu kommen geschätzte 110.000 Todesfälle/Jahr aufgrund von Opioid-Überdosierungen allein in den USA.
Da die Natriumkanäle 1.6 bis 1.8 eine zentrale Rolle beim »Feuern« der Aktionspotenziale bei der Nozizeption spielen, ist es per se erfolgversprechend, diese Kanäle zu blockieren. Da der Nav1.6-Kanal sowohl im peripheren als auch im zentralen Nervensystem vorkommt, ist er aufgrund der zu erwartenden kognitiven, motorischen und sensorischen Nebenwirkungen wenig geeignet für die Schmerztherapie.
Mit einer Art dynamischer Patch-Clamp-Methode, die elektronisch den Nav1.7-Strom an sensorischen Neuronen entfernen kann, konnte sichergestellt werden, dass die Nav1.7-Kanäle essenziell für die Schmerzweiterleitung sind, also ihre Inhibierung zur Schmerzbehandlung herangezogen werden kann. Das Arylsulfonamid PF-05089771 (Abbildung 3), das strukturelle Ähnlichkeit zum Nav1.5-Blocker NBI-9321352 hat, zeigte zwar eine große Selektivität zum Nav1.7, hatte aber in klinischen Studien nur wenig Effekt, wahrscheinlich weil es sehr stark an Plasmaprotein gebunden wird.
In Tierversuchen wurde zudem die analgetische Wirkung des in der Peripherie wirkenden Sulfonamids GEN-3565 (Abbildung 3) gezeigt. Der Wirkstoff wies zudem günstige pharmakokinetische und pharmakodynamische Eigenschaften auf (12). GEN-3565 bindet an die VSD4-Domäne (13).
Auch Tetrodotoxin und Saxitoxin, beide gekennzeichnet durch (sehr basische und damit protonierte) Guanidinium-Strukturelemente, sind potente Blocker des Nav1.7-Kanals, zeigen aber Nebenwirkungen am autonomen Nervensystem, zum Beispiel Hypotonie. Alle Substanzen sind noch weit von klinischen Untersuchungen entfernt (5).
Im Januar 2025 wurde das oral verfügbare Suzetrigin (Journavx®) von Vertex (VX-158, Abbildung 4) von der FDA zugelassen. Es ist der erste Nav-Kanalblocker, mit dem akute und neuropathische Schmerzen gut behandelt werden können. Es stabilisiert sehr selektiv den geschlossenen Nav1.8-Kanal durch Bindung an VSD2; zum Vergleich: Lidocain blockiert alle offenen Natriumkanäle!
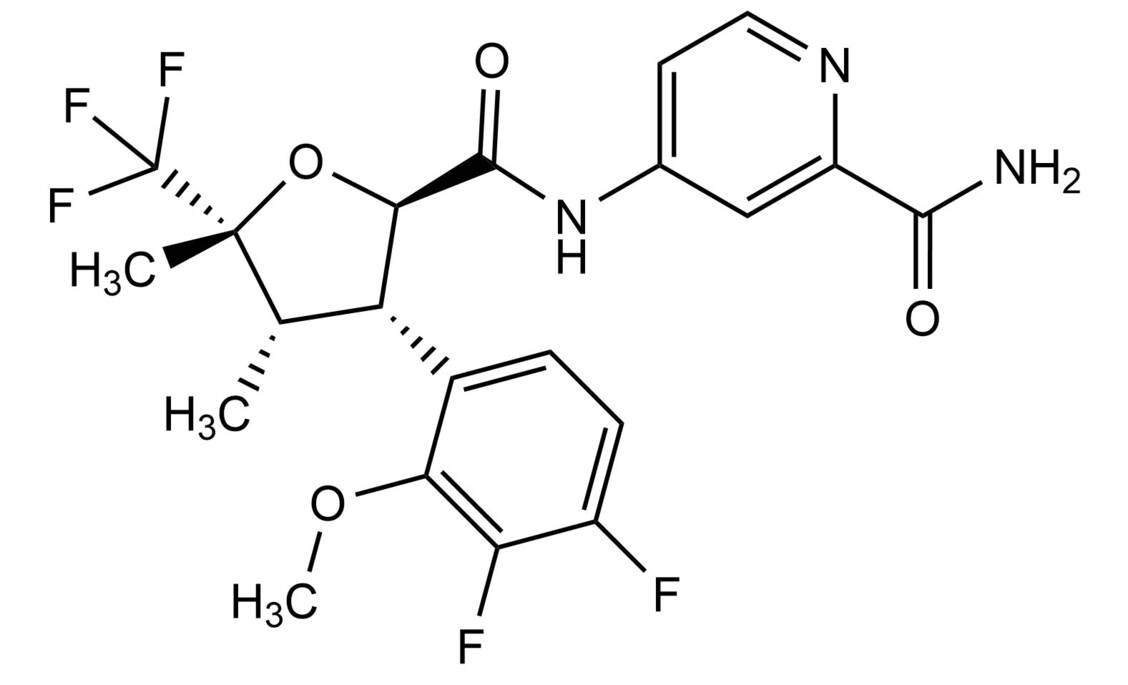
Abbildung 4: Nav1.8-Inhibitor Suzetrigin / © PZ/Wurglics
Dieser neue allostere Wirkmechanismus führt zu einer tonischen Inhibition von Nav1.8, der aufgrund seiner hohen »Feuerfrequenz« am stärksten an der Schmerzweiterleitung beteiligt ist. Seine Blockade reduziert effektiv das Schmerzsignal in peripheren sensorischen Neuronen (14). Aufgrund der Selektivität hat Suzetrigin keine kardiovaskulären (Blutdruck, QT-Zeit-Verlängerung) und respiratorischen Nebenwirkungen. Allerdings können Juckreiz, Hautausschlag, Muskelkrämpfe und erhöhte Kreatin-Phosphokinase-Werte auftreten (14, 15).
Nach einer Initialdosis von 100 mg und einer Folgedosierung von 50 mg alle zwölf Stunden war die Wirkung von Suzetrigin bei mittelschweren und schweren akuten Schmerzen signifikant besser als Placebo und vergleichbar mit Hydrocodon (NAVIGATE2-Studie). Bei neuropathischen Schmerzen, verursacht durch eine periphere diabetische Neuropathie, wurden eine signifikante Reduktion bei einer relativ geringen Dosis von etwa 50 mg/d erzielt. Aus den Zusammenfassungen aller bisher publizierten Phase-II- und -III-Studien (15) geht allerdings hervor, dass nur das 100/50-mg-Schema eine zuverlässige Schmerzreduktion bewirkt.

Das oral verfügbare Suzetrigin ist der erste Nav-Kanalblocker zur Therapie von akuten und neuropathischen Schmerzen. / © Shutterstock/Benevolente82
Suzetrigin wird im Wesentlichen von CYP3A metabolisiert, wobei es einen aktiven Metaboliten (M6-SUZ) gibt, der wahrscheinlich die O-desmethylierte Form ist. Die Strukturen der Metaboliten sind bisher nicht publiziert. Da die Substanz selbst und auch der Metabolit CYP3A-Substrate sind, sollte Suzetrigin nicht zusammen mit CYP3A-Inhibitoren appliziert werden, da es ansonsten zu überhöhten Konzentrationen und damit Nebenwirkungen kommt. Umgekehrt sollten auch CYP3A-Induktoren vermieden werden. Deshalb sollte Suzetrigin nüchtern oder mit einem Zwei-Stunden-Abstand zum Essen eingenommen und Grapefruitsaft gänzlich vermieden werden.
Vorsicht ist geboten bei der Kontrazeption. Es sollten zusätzlich eine nicht hormonelle Verhütungsmethode vorgenommen werden.
Die pharmakokinetischen Daten sind in Tabelle 2 zusammengefasst. Suzetrigin wird in der Leber metabolisiert und zu etwa 50 Prozent über den Stuhl ausgeschieden, dabei zu 9 Prozent unverändert. 44 Prozent werden mit dem Urin eliminiert.
| Pharmakokinetik | Suzetrigin | M6-Suzetrigin |
|---|---|---|
| Dosis (mg) | 100 beziehungsweise 50 | — |
| tmax (h) | 3 | 8 bis 10 |
| Cmax (µg/L) | 0,62 | 11,5 |
| AUC (µg/mL) | 11,5 | 34,7 |
| t1/2 (h) | 23 | 33 |
| Verteilungsvolumen (L) | 495 | — |
| Plasmaproteinbindung (Prozent) | 99 | 96 |


