 |  | Ulrike Holzgrabe |
 |
02.11.2025 08:00 Uhr |

Natriumkanäle spielen eine vielfältige Rolle in der (Patho-)Physiologie vieler Erkrankungen, unter anderem bei epileptischen Anfällen. / © Shutterstock/Madrolly
Von Lokalanästhetika und Antiarrhythmika sowie manchen Antiepileptika wissen wir seit Langem, dass sie mit den spannungsabhängigen Natriumkanälen in Wechselwirkung treten, um den Natriumeinstrom zu blockieren und damit ihre Wirkung zu entfalten. Bislang sind neun Kanaltypen bekannt, die an verschiedenen Organsystemen lokalisiert sind und eine ähnliche Grundstruktur haben – mit geringfügigen, aber wichtigen Unterschieden. Diese Details kann man zur Entwicklung selektiver Arzneistoffe nutzen. Die Struktur der Natriumkanäle, ihre Aktivierung und die Wirkweise der interagierenden Arzneistoffe werden hier dargestellt.
Der Ablauf eines Aktionspotenzials ist mit Schwellenspannung, Depolarisation (Na⁺-Einstrom), Repolarisation (K⁺-Ausstrom), Hyperpolarisation und wieder Ruhepotenzial schon lange bekannt. Dagegen wurden die strukturellen Details der Natriumkanäle und ihre Funktionsweise erst in den letzten Jahren durch kryo-elektronenmikroskopische und Moleküldynamik-Untersuchungen erforscht.
Die Natriumkanäle (Nav) gehören zur Superfamilie der spannungsabhängigen (voltage-gated) Kationenkanäle, wie auch Kv und Cav-Kanäle.
Prinzipiell bestehen die komplex aufgebauten, aus etwa 2000 Aminosäuren bestehenden Natriumkanäle aus einer α-Einheit, die aus vier miteinander verbundenen, nicht identischen, aber homologen Domänen (DI bis DIV) besteht. Jede Domäne besteht wiederum aus sechs miteinander verbundenen transmembranären Segmenten (S1 bis S6), wobei S1 bis S4 spannungsempfindliche Module (voltage-sensing domains VSD1 bis VSD4) sind und S5 und S6 eine zentrale Pore bilden (Porenmodul PM) (1). Dabei sind S4 und S5 über einen Linker miteinander verbunden. Die Abbildung 1 zeigt den komplizierten Aufbau anschaulich. S5 und S6 sind über einen extrazellulären Haarnadel-Loop miteinander verbunden, der den Ionenkanälen den Namen P-Loop-Kanäle gegeben hat.
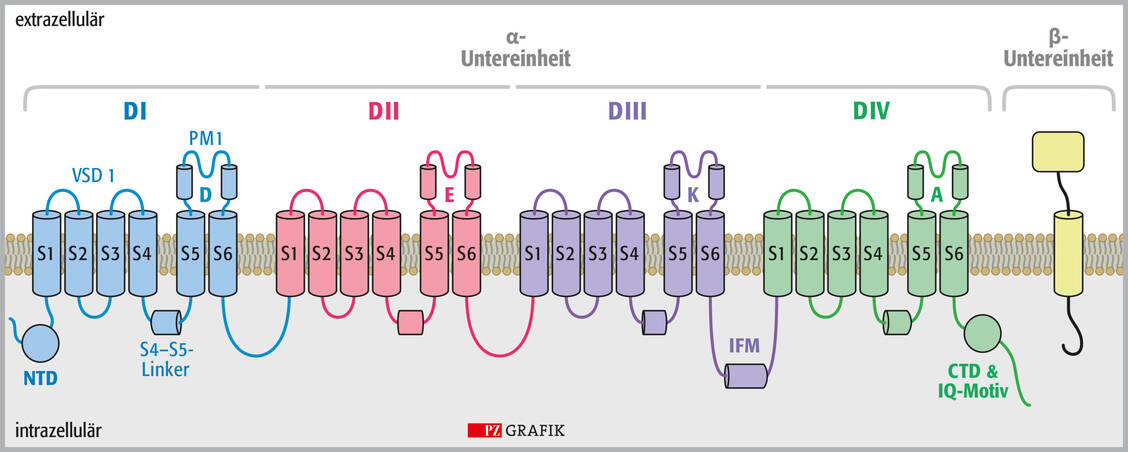
Abbildung 1A: Schematische Darstellung des humanen Nav-Kanals; links die α-Untereinheit mit den Domänen DI bis DIV und den Segmenten S1 bis S6; rechts (gelb gezeichnet) die β-Untereinheit. CTD: Carboxyl-terminale Domäne; D: Domäne; IFM: Fast inactivation peptide, Peptid, das für die schnelle Inaktivierung verantwortlich ist; IQ-Motiv: Calmodulin-Bindemotiv; PM: Porenmodul; NTD: N-terminale Domäne; S: transmembranäres Segment; VSD: spannungsempfindliche Module, voltage-sensor domain / © PZ/Stephan Spitzer
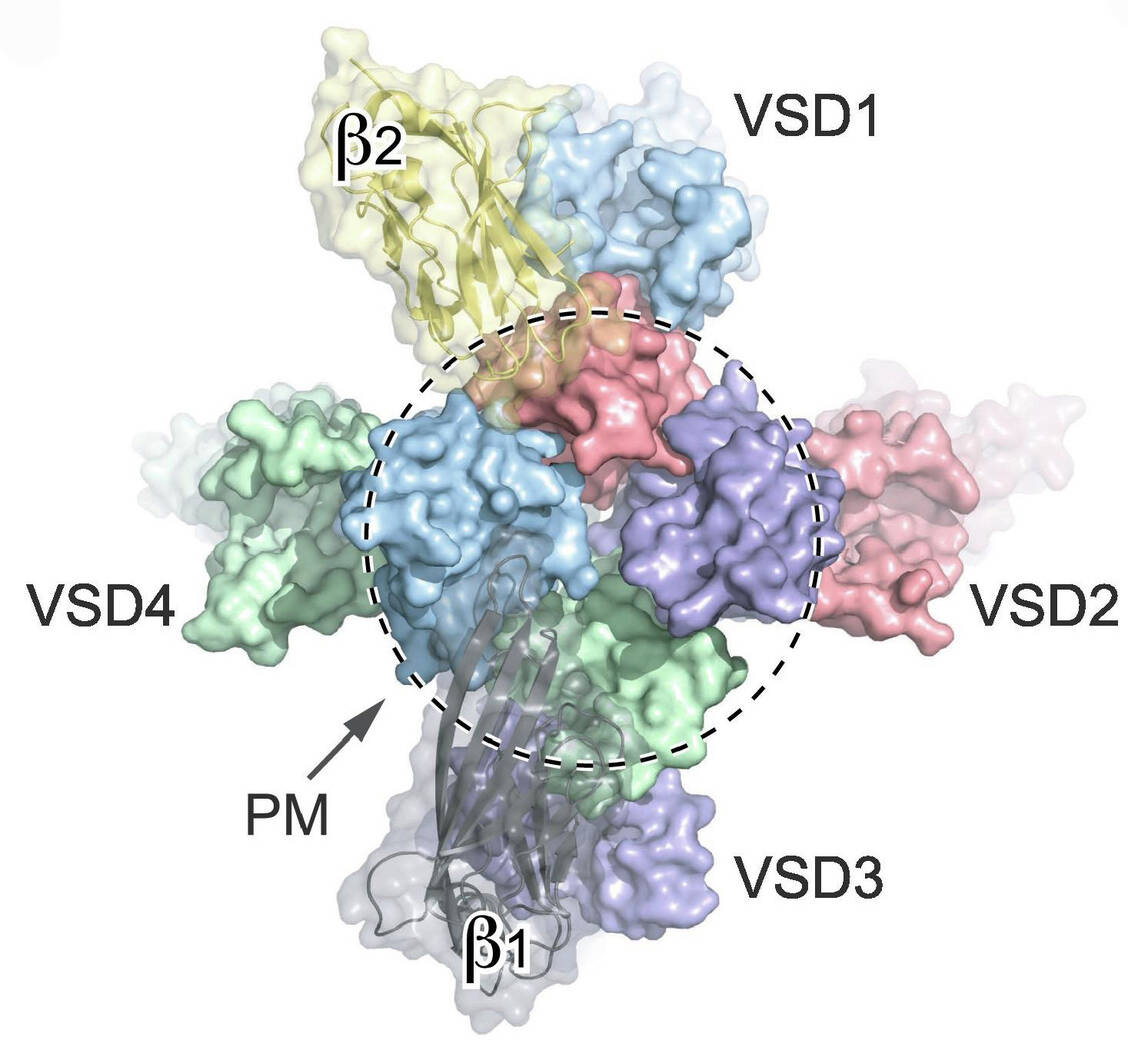
Abbildung 1B: Extrazellulärer Blick auf den humanen Nav1.7-Kanal (Protein Data Bank, PDB: 6J8J); in der Mitte die Pore (PM) und an den vier Ecken die spannungsempfindlichen Domänen (VSD) und die β-Untereinheit; alle Teile haben dieselben Farben wie in Abbildung 1A. / © PDB
Die vier spannungsempfindlichen Module sind an den Ecken der zentralen Pore lokalisiert. Die VSD4-Untereinheit weist ein Motiv mit positiv geladenen Argininen auf. Im Ruhezustand des Kanals sind diese positiv geladenen Aminosäuren durch das negative Membranpotenzial einwärtsgerichtet. Kommt es zur Depolarisation, bewegen sich diese Aminosäuren nach außen und bilden mit negativ geladenen Aminosäuren in den S1- bis S3-Untereinheiten Ionenpaare, sodass der Ionenkanal geöffnet wird.
An diesem Prozess sind die Porenmodule S5 und S6 beteiligt (Abbildung 1). Diese bestehen aus
Wie gelangen nun Arzneistoffe wie Lokalanästhetika oder Antiarrhythmika in den Natriumkanal, um ihn blockieren zu können? Zwei Wege sind den sogenannten Porenblockern möglich.
In beiden Fällen müssen die Arzneistoffe, zum Beispiel Lokalanästhetika, die lipophile Membran passieren, also in einer lipophilen Form vorliegen. In beiden Fällen verschließen sie Teile des Kanals und eine Passage der Natriumionen ist nicht mehr möglich oder sie stabilisieren den inaktiven Zustand oder beides. In der zentralen Kavität liegen die Arzneistoffe positiv geladen vor und interagieren mit Tyrosin-Resten im Natriumkanal (2). Um für beide Prozesse geeignet zu sein, müssen sie sowohl positiv geladen (protoniert) als auch neutral (unprotoniert) sein können. Dazu müssen die sekundären oder tertiären Amine der Lokalanästhetika einen pKa zwischen 7,5 und 9,0 haben (Abbildung 2).

Abbildung 2: Nach dem Löfgren-Schema besteht ein Lokalanästhetikum aus einem lipophilen, meist aromatischen Molekülteil, der über eine kurze Kohlenstoffkette mit einem hydrophilen basischen Teil verknüpft ist. Hier sind die Ladungszustände der Lokalanästhetika ergänzt. / © PZ/Stephan Spitzer
Die neutralen Stoffe Carbamazepin und Lamotrigin komplexieren vermutlich ein Natriumion, um geladen zu sein (3).
Die Frage stellt sich, welchen Weg welcher Arzneistoff nimmt und wie er jeweils bindet. Obgleich es viele Moleküldynamikrechnungen mit verschiedenen Verbindungen gibt (4), sind keine klaren Regeln erkennbar. Neutrale Arzneistoffe wie Benzocain und Phenytoin scheinen »gerne« Interdomänen-Fenster aufgrund ihrer kleinen Größe zu nutzen. Dagegen können Lokalanästhetika wie Lidocain oder Etidocain beide Wege nehmen.
Lokalanästhetika werden häufig bei kleinen Operationen oder Zahnbehandlungen appliziert. Sie sollen dabei lokal wirken und nicht in den Kreislauf gelangen. / © Shutterstock/Ocskay Mark
Zudem gibt es offensichtlich mehrere Bindemodi. Die neutrale Aminseitenkette kann die Fenster verschließen und damit den Natriumkanal verengen, sodass Natriumionen nicht mehr durch den Selektivitätsfilter gelangen. Alternativ kann ein positiv geladenes Lokalanästhetikum (Abbildung 2) den Selektivitätsfilter verschließen – mit dem gleichen Effekt. Dabei spielen π-Kation- und π-π-Wechselwirkungen offensichtlich eine Rolle, wie Mutationsstudien gezeigt haben. Auch das Aktivierungstor kann der Bindeort sein. Bei allem spielt natürlich noch der Ladungszustand des Kanals eine Rolle, für den es aber auch mehrere Hypothesen gibt.
Hiermit stellt sich eine weitere Frage: Reicht das bisherige Wissen, um rational neue Wirkstoffe zu designen, die gezielt mit einem der Natriumkanäle wechselwirken?
Erregbare Zellen, die von spannungsabhängigen Natriumkanälen reguliert werden, finden sich in Neuronen, Kardiomyozyten und Muskelzellen. Natriumkanäle ähneln sich zwar vom generellen Aufbau und der allgemeinen Funktionsweise her, aber die neun Kanalsubtypen sind strukturell unterschiedlich und ihre Erregung (Na⁺-Einstrom) hat unterschiedliche Funktionen. Umgekehrt formuliert: Ihre Störung ruft unterschiedliche Krankheitsbilder hervor.
Während Nav1.1, 1.2 und 1.6 bei zu hoher Feuerfrequenz epileptische Anfälle hervorrufen können, löst eine Übererregung von Nav1.4 Myotonie (vermehrte Muskelspannung) und von Nav1.5 Arrhythmien am Herzen aus. Nav1.7 bis 1.9 steuern die Schmerzverarbeitung (Tabelle 1).
| Natriumkanal | Lokalisation | Erkrankungen bei Dysfunktion | Inhibitoren |
|---|---|---|---|
| Nav1.1 | ZNS | Epilepsie, Autismus | |
| Nav1.2 | ZNS | Epilepsie, Autismus | |
| Nav1.4 | Skelettmuskel | Myotonie, Paralyse | |
| Nav1.5 | Herzmuskel | langes QT-Syndrom, Arrhythmien | |
| Nav1.6 | ZNS/PNS | Epilepsien, Absencen | NBI-921352, Prax562 |
| Nav1.7 | PNS | Schmerz | PF-05089771, GEN-3565 |
| Nav1.8 | PNS | Schmerz | Suzetrigin |
| Nav1.9 | PNS | Schmerz |
Alle bisher bekannten Arzneistoffe, die mit Natriumkanälen interagieren, blockieren mehr oder weniger unselektiv alle Kanäle, wobei Lokalanästhetika sowohl tonisch als auch use-dependent in neutralem und geladenem Zustand des Kanals agieren. Moleküldynamik-Simulationen haben ergeben, dass die neutralen Wirkstoffe Carbamazepin und Lamotrigin sowie Lidocain im Zentrum der großen Kavität binden, wenn auch an verschiedenen Stellen (6). Solche mittels Moleküldynamik erzeugten Ergebnisse erzielt man auch für andere neutrale Antiepileptika.
Da Lokalanästhetika Schmerz lokal unterdrücken sollen, zum Beispiel bei kleinen Operationen oder Zahnbehandlungen, werden sie auch lokal in der Peripherie appliziert und sollten dabei nicht in den Kreislauf gelangen. Das bedeutet, dass eine selektive Blockade nur von Nav1.7 bis 1.9 gar nicht notwendig ist. Dagegen ist bei der Behandlung von Arrhythmien, Epilepsien oder Schmerz eine gewisse Subtyp-Selektivität zu einzelnen Natriumkanälen wünschenswert.
Zur Unterdrückung von epileptischen Anfällen sollen die Natriumkanäle Nav1.1, 1.2 und/oder 1.6 blockiert werden (5) (Tabelle 1). Es ist bisher nicht gelungen, Nav1.1 oder 1.2 selektiv zu blockieren, weder mit kleinen Molekülen noch mit anderen Strategien.

Epileptische Anfälle können in jedem Alter auftreten. Die entwicklungsbedingte epileptische Enzephalopathie bei Kleinkindern geht mit einer anomalen Überaktivität des Nav1.6-Kanals einher. / © Shutterstock/vasara
Im Unterschied dazu gibt es für die im frühkindlichen Alter auftretende »entwicklungsbedingte (developmental) epileptische Enzephalopathie (DEE)« zwei Phase-II-Kandidaten. Diese Epilepsieform geht mit einer anomalen Überaktivität des Nav1.6 (»Gain-of-Function«-Mutante des Nav1.6-Kanals) einher. Die seltene (1 bis 3 Patienten pro 100.000), aber schwere neuropädiatrische Erkrankung ist durch psychomotorische und kognitive Störungen sowie Verhaltensauffälligkeiten gekennzeichnet (7). Das Sulfonamid NBI-921352 (Abbildung 3) verhindert Krampfanfälle in geringeren zerebralen und Plasma-Konzentrationen im Gehirn als diese bei einer Therapie mit den Antiepileptika Carbamazepin, Phenytoin oder Lacosamid notwendig sind (8).
Die hoch fluorierte Verbindung Prax562 (Relutrigin, Abbildung 3) unterdrückt den Ausbruch des Aktionspotenzials und reduziert dieses; sie wirkt bei DEE-Patienten ebenso gut wie Carbamazepin und ist Lamotrigin überlegen (9). In die multizentrische doppelblinde Phase-II-Studie EMBOLD wurden 16 Patienten zwischen 2 und 18 Jahren einbezogen. Relutrigin wurde gut vertragen und konnte die Zahl der Krampfanfälle nach 16 Wochen um 46 Prozent und nach elf Monaten um 90 Prozent reduzieren (10). Dieses sehr positive Ergebnis, das als Durchbruch in der Therapie der DEE gefeiert wird, muss nun in Phase-III-Studien, für die gerade Patienten rekrutiert werden, überprüft werden.
Auch Cannabidiol hemmt den Nav1.6-Kanal und ist effektiv bei Arzneistoff-resistenten Krampfanfällen. Allerdings beeinflusst es eine ganze Reihe anderer Zielstrukturen (5), was zu Off-Target-Effekten führt.
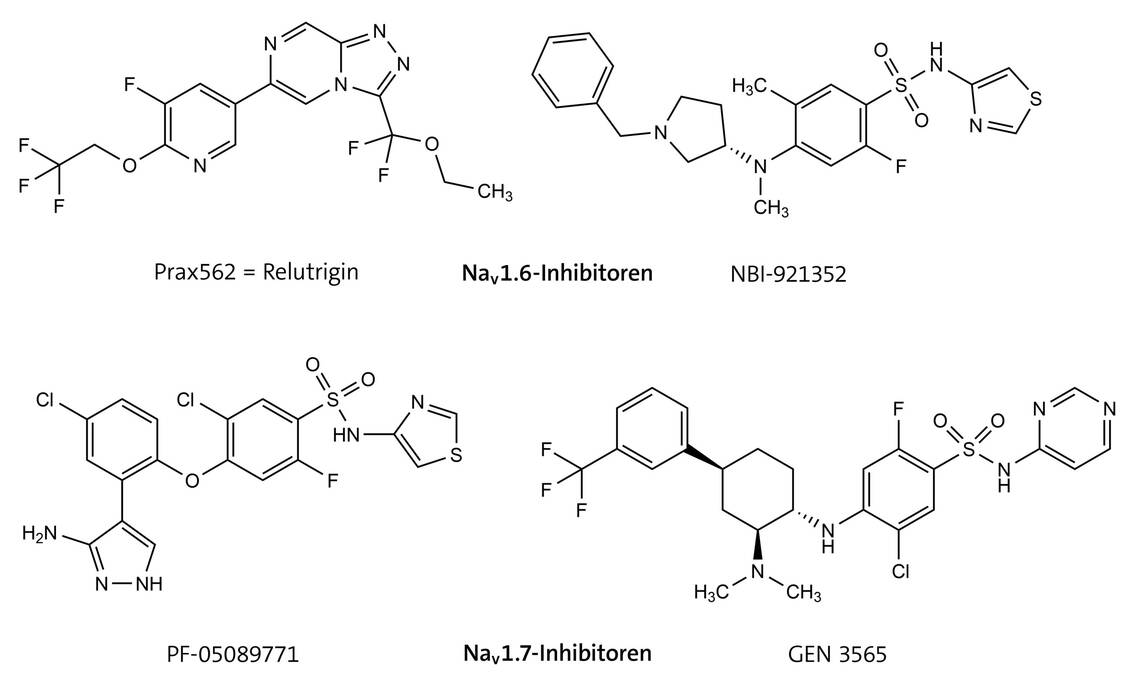
Abbildung 3: Strukturen der Nav1.6-Inhibitoren Prax562 (Relutrigin) und NBI-921352 sowie der Nav1.7-Inhibitoren PF-05089771 und GEN-3565 / © PZ/Wurglics
Funktionsstörungen, die durch den Nav1.4 verursacht werden, sind sehr selten. Häufig ist ein Arginin-Rest gegen ein Cystein, Histamin oder Prolin ausgetauscht. Meist wird eine beeinträchtigte oder verzögerte Relaxation der Skelettmuskulatur beobachtet. Die dadurch verursachten Myopathien werden mit Antiarrhythmika oder Antiepileptika wie Mexiletin, Ranolazin oder Lamotrigin behandelt. Weitere bekannte Arzneistoffe sind in klinischen Prüfungen für die neue Indikation (»Repurposing«).
Erwähnt sei, dass das bei Morbus Parkinson eingesetzte Safinamid neben der MAO-B-Hemmung auch den Nav1.4-Kanal und damit die anomale Muskelübererregbarkeit sowie die damit einhergehende Myotonie hemmt (11).
Im Unterschied zum Verlauf des Aktionspotenzials im zentralen und im peripheren Nervensystem sowie im Skelettmuskel ist der Verlauf am Herzen durch eine Plateauphase nach der Depolarisation nach Natriumeinstrom charakterisiert. Diese wird durch einen Calciumeinstrom unterhalten, bevor dann ein Kaliumausstrom zur Repolarisation bis zum Ruhepotenzial folgt. Zur Behandlung einer Arrhythmie können alle Ionenströme durch Arzneistoffe beeinflusst werden.
Bestimmte Natriumkanäle am Herzen sind an der Entstehung von Arrhythmien beteiligt. / © Shutterstock/Liya Graphics
Antiarrhythmika der Klasse 1 beeinflussen den Natriumeinstrom am Nav1.5-Kanal und werden bei der Behandlung des langen QT-Syndroms (LQT) eingesetzt (verlängerte QT-Zeit). Es gibt verschiedene LQT-Varianten. Beispielsweise destabilisiert LQT3 die schnelle Inaktivierung des Kanals, was zu einem persistierenden Na⁺-Einwärtsstrom führt und damit zu einer verspäteten Repolarisation.
LQT-Syndrome werden häufig mit Betablockern behandelt, wobei der Erfolg nicht garantiert ist. Die Blockade der Natriumkanäle scheint effektiver zu sein. Mexiletin, ein Klasse-1b-Antiarrhythmikum, hat eine hohe Affinität zu überaktiven Nav1.5 und bewirkt eine Use-dependent-Blockade. Flecainid, ein Klasse-1c-Antiarrhythmikum, blockiert den Kanal im offenen Zustand. Ranolazin, ebenfalls ein Klasse-1c-Antiarrhythmikum, hat eine hohe Affinität zum persistierenden Natriumeinstrom in den Arterien.
Alle Blocker sind nicht ideal. Jedoch kam die Entwicklung von neuen Antiarrhythmika nach der Einführung von Dronedaron 2009 zum Stillstand, was aufgrund schwerwiegender Nebenwirkungen zwar verständlich, aber sehr bedenklich ist (5).
Die derzeitige Behandlung von Schmerzen, insbesondere von chronischen Schmerzen, mit nicht steroidalen Antirheumatika (NSAR) oder Opioiden ist durch schwere Nebenwirkungen sowie Suchtpotenzial geprägt. Hinzu kommen geschätzte 110.000 Todesfälle/Jahr aufgrund von Opioid-Überdosierungen allein in den USA.
Da die Natriumkanäle 1.6 bis 1.8 eine zentrale Rolle beim »Feuern« der Aktionspotenziale bei der Nozizeption spielen, ist es per se erfolgversprechend, diese Kanäle zu blockieren. Da der Nav1.6-Kanal sowohl im peripheren als auch im zentralen Nervensystem vorkommt, ist er aufgrund der zu erwartenden kognitiven, motorischen und sensorischen Nebenwirkungen wenig geeignet für die Schmerztherapie.
Mit einer Art dynamischer Patch-Clamp-Methode, die elektronisch den Nav1.7-Strom an sensorischen Neuronen entfernen kann, konnte sichergestellt werden, dass die Nav1.7-Kanäle essenziell für die Schmerzweiterleitung sind, also ihre Inhibierung zur Schmerzbehandlung herangezogen werden kann. Das Arylsulfonamid PF-05089771 (Abbildung 3), das strukturelle Ähnlichkeit zum Nav1.5-Blocker NBI-9321352 hat, zeigte zwar eine große Selektivität zum Nav1.7, hatte aber in klinischen Studien nur wenig Effekt, wahrscheinlich weil es sehr stark an Plasmaprotein gebunden wird.
In Tierversuchen wurde zudem die analgetische Wirkung des in der Peripherie wirkenden Sulfonamids GEN-3565 (Abbildung 3) gezeigt. Der Wirkstoff wies zudem günstige pharmakokinetische und pharmakodynamische Eigenschaften auf (12). GEN-3565 bindet an die VSD4-Domäne (13).
Auch Tetrodotoxin und Saxitoxin, beide gekennzeichnet durch (sehr basische und damit protonierte) Guanidinium-Strukturelemente, sind potente Blocker des Nav1.7-Kanals, zeigen aber Nebenwirkungen am autonomen Nervensystem, zum Beispiel Hypotonie. Alle Substanzen sind noch weit von klinischen Untersuchungen entfernt (5).
Im Januar 2025 wurde das oral verfügbare Suzetrigin (Journavx®) von Vertex (VX-158, Abbildung 4) von der FDA zugelassen. Es ist der erste Nav-Kanalblocker, mit dem akute und neuropathische Schmerzen gut behandelt werden können. Es stabilisiert sehr selektiv den geschlossenen Nav1.8-Kanal durch Bindung an VSD2; zum Vergleich: Lidocain blockiert alle offenen Natriumkanäle!
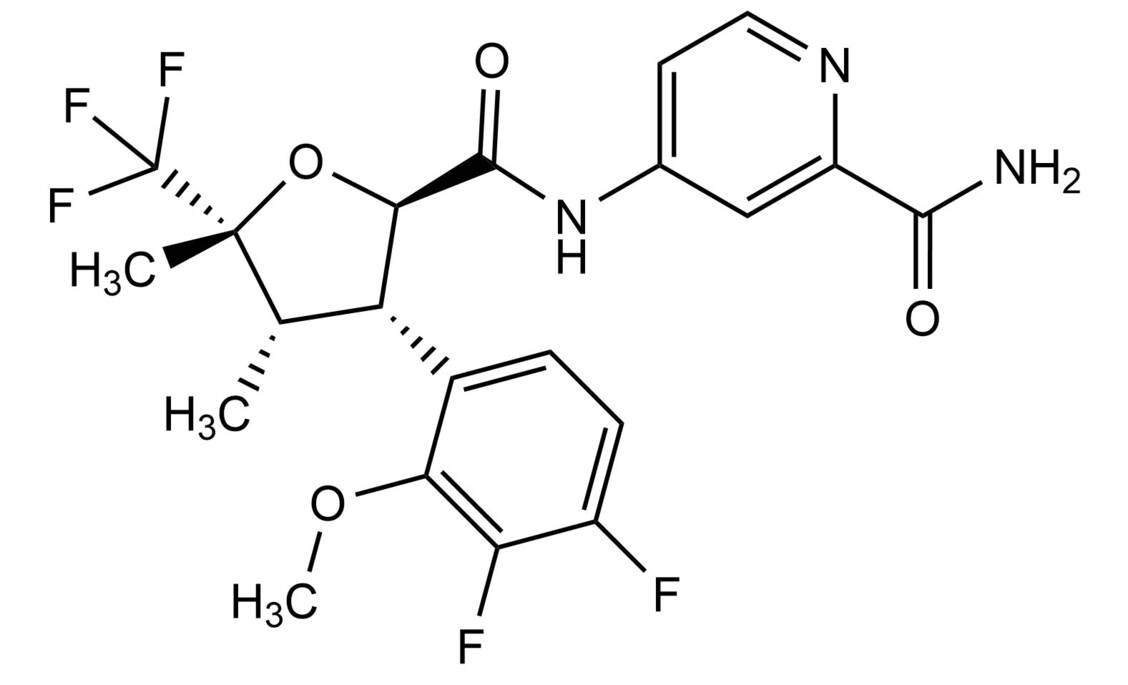
Abbildung 4: Nav1.8-Inhibitor Suzetrigin / © PZ/Wurglics
Dieser neue allostere Wirkmechanismus führt zu einer tonischen Inhibition von Nav1.8, der aufgrund seiner hohen »Feuerfrequenz« am stärksten an der Schmerzweiterleitung beteiligt ist. Seine Blockade reduziert effektiv das Schmerzsignal in peripheren sensorischen Neuronen (14). Aufgrund der Selektivität hat Suzetrigin keine kardiovaskulären (Blutdruck, QT-Zeit-Verlängerung) und respiratorischen Nebenwirkungen. Allerdings können Juckreiz, Hautausschlag, Muskelkrämpfe und erhöhte Kreatin-Phosphokinase-Werte auftreten (14, 15).
Nach einer Initialdosis von 100 mg und einer Folgedosierung von 50 mg alle zwölf Stunden war die Wirkung von Suzetrigin bei mittelschweren und schweren akuten Schmerzen signifikant besser als Placebo und vergleichbar mit Hydrocodon (NAVIGATE2-Studie). Bei neuropathischen Schmerzen, verursacht durch eine periphere diabetische Neuropathie, wurden eine signifikante Reduktion bei einer relativ geringen Dosis von etwa 50 mg/d erzielt. Aus den Zusammenfassungen aller bisher publizierten Phase-II- und -III-Studien (15) geht allerdings hervor, dass nur das 100/50-mg-Schema eine zuverlässige Schmerzreduktion bewirkt.

Das oral verfügbare Suzetrigin ist der erste Nav-Kanalblocker zur Therapie von akuten und neuropathischen Schmerzen. / © Shutterstock/Benevolente82
Suzetrigin wird im Wesentlichen von CYP3A metabolisiert, wobei es einen aktiven Metaboliten (M6-SUZ) gibt, der wahrscheinlich die O-desmethylierte Form ist. Die Strukturen der Metaboliten sind bisher nicht publiziert. Da die Substanz selbst und auch der Metabolit CYP3A-Substrate sind, sollte Suzetrigin nicht zusammen mit CYP3A-Inhibitoren appliziert werden, da es ansonsten zu überhöhten Konzentrationen und damit Nebenwirkungen kommt. Umgekehrt sollten auch CYP3A-Induktoren vermieden werden. Deshalb sollte Suzetrigin nüchtern oder mit einem Zwei-Stunden-Abstand zum Essen eingenommen und Grapefruitsaft gänzlich vermieden werden.
Vorsicht ist geboten bei der Kontrazeption. Es sollten zusätzlich eine nicht hormonelle Verhütungsmethode vorgenommen werden.
Die pharmakokinetischen Daten sind in Tabelle 2 zusammengefasst. Suzetrigin wird in der Leber metabolisiert und zu etwa 50 Prozent über den Stuhl ausgeschieden, dabei zu 9 Prozent unverändert. 44 Prozent werden mit dem Urin eliminiert.
| Pharmakokinetik | Suzetrigin | M6-Suzetrigin |
|---|---|---|
| Dosis (mg) | 100 beziehungsweise 50 | — |
| tmax (h) | 3 | 8 bis 10 |
| Cmax (µg/L) | 0,62 | 11,5 |
| AUC (µg/mL) | 11,5 | 34,7 |
| t1/2 (h) | 23 | 33 |
| Verteilungsvolumen (L) | 495 | — |
| Plasmaproteinbindung (Prozent) | 99 | 96 |
Die selektive Blockade der einzelnen spannungsabhängigen Natriumkanäle – Präzisionsmedizin genannt – birgt offenbar ein großes therapeutisches Potenzial, das es zu nutzen gilt. Suzetrigin ist ein erster Schritt. Weitere Arzneistoffe sind in der klinischen Pipeline, zum Beispiel zur Behandlung von Epilepsie und Schmerz (Nav1.8-Blocker). Viele neue Moleküle, die mithilfe der Strukturkenntnis der Kanäle und der Kryo-Elektronenmikroskopie designt wurden, müssen »nur« noch optimiert werden.
Ulrike Holzgrabe studierte Chemie und Pharmazie in Marburg und Kiel und habilitierte sich in Pharmazeutischer Chemie in Kiel. Nach mehrjähriger Professorentätigkeit in Bonn ist sie seit April 1999 als Professor in Würzburg tätig, seit Mitte 2022 als Emerita. Professor Holzgrabe war von 2018 bis 2021 Vizepräsidentin der Universität Würzburg. In vielfältigen Positionen arbeitete sie am Deutschen und Europäischen Arzneibuch am BfArM und EDQM mit. Seit vielen Jahren forscht sie auf dem Gebiet der Antibiotika und der Analytik.


