 |  | Bettina Wick-Urban |
 |
08.02.2026 08:00 Uhr |
Um bereits bei der Zulassung eine größtmögliche Transparenz zu schaffen, müssen seit 2016 die Arzneimittelhersteller bereits mit Einreichen des Zulassungsantrags für ein neues Medikament oder bei einer Zulassungserweiterung eine strukturierte und systematische Nutzen-Risiko-Bewertung in der klinischen Zusammenfassung bei den Behörden vorlegen (12, 13). Voraussetzung für die Erteilung der Zulassung ist ein positives Nutzen-Risiko-Verhältnis, das heißt: Der Nutzen des Arzneimittels muss für eine bestimmte Indikation in einer bestimmten Patientengruppe die Risiken überwiegen.
Was beinhaltet die Bewertung? Das Nutzen-Risiko-Verhältnis wird in einer strukturierten wissenschaftlichen Bewertung ermittelt, die quantitative und qualitative Daten integriert. Es gibt verschiedene Ansätze von Zulassungsbehörden und wissenschaftlichen Organisationen, die im Wesentlichen auf drei Säulen beruhen: dem therapeutischen Kontext, den klinisch relevanten Wirksamkeitsparametern und Risiken sowie der Risikooptimierung (Grafik) (13).
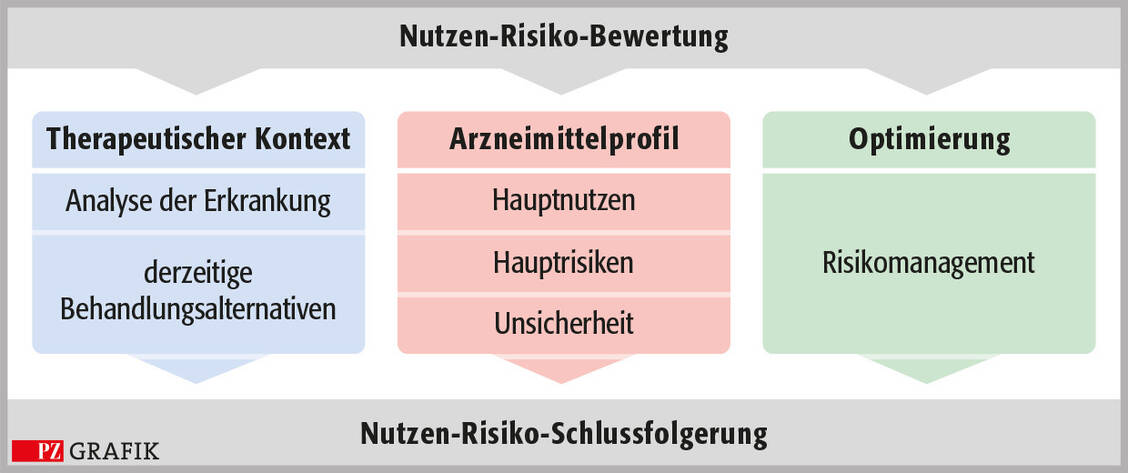
Grafik: Drei Säulen einer strukturierten Nutzen-Risiko-Bewertung; modifiziert nach: Benefit-Risk-Balance for Medicinal Products. CIOMS Working Group report. Geneva, 2025 / © PZ/Stephan Spitzer
Der therapeutische Kontext beschreibt die Erkrankung, deren Symptome, Schweregrad und Häufigkeit sowie – wichtig aus der Perspektive des Patienten – die damit verbundenen Beeinträchtigungen der Lebensqualität, aber auch mögliche Auswirkungen auf das Gesundheitssystem, zum Beispiel erhöhte Kosten durch Operationen oder Krankenhausaufenthalte. Der zweite wichtige Aspekt im therapeutischen Kontext sind die Analyse des Nutzens und der Risiken von alternativen Behandlungsmöglichkeiten sowie die Überlegung, welche Therapielücken aus Sicht der Verschreiber und der Patienten bestehen.
Den Kern der Bewertung stellt die Analyse des Profils des neuen Arzneimittels dar (Grafik, Mitte), das heißt die klinisch relevanten Wirksamkeitsparameter und die positiven Einflüsse auf die gesundheitsbezogene Lebensqualität aus der Perspektive des Patienten sowie andererseits die schwerwiegenden Risiken, die das Nutzen-Risiko-Verhältnis maßgeblich beeinflussen, basierend auf den Daten der (nicht) klinischen Studien. Zum Zeitpunkt der Einreichung des Zulassungsantrags liegen natürlich nur begrenzte Daten vor, zum Beispiel zur Langzeitwirksamkeit, zu seltenen schwerwiegenden Nebenwirkungen oder zu Nebenwirkungen, die erst mit einer gewissen Latenz auftreten, oder zu Patientengruppen, die in die klinischen Studien nicht eingeschlossen wurden. Daher ist der Antragsteller verpflichtet, einen Plan vorzulegen, wie diese Lücken nach der Zulassung geschlossen werden.
Die dritte wichtige Säule der Bewertung ist die Risikooptimierung. Damit ein Arzneimittel ein möglichst günstiges Nutzen-Risiko-Verhältnis hat, muss der Antragsteller die Risikominimierungsmaßnahmen beschreiben. Zu den Routinemaßnahmen gehören zum Beispiel die Verschreibungspflicht, die Packungsgröße oder die Verpackung des Arzneimittels (kindersichere Verschlüsse).
Die wichtigsten Maßnahmen sind in der Fachinformation und der Packungsbeilage festgehalten. Darin wird beschrieben, für welche Erkrankung und welche Patienten ein Produkt in welcher Dosierung zugelassen ist. Weiterhin sind die Kontraindikationen, Wechselwirkungen und Arzneimittelnebenwirkungen beschrieben. Wichtig sind auch die besonderen Warnhinweise und Vorsichtsmaßnahmen für die Anwendung. Hier sind unter anderem schwerwiegende Nebenwirkungen oder Umstände aufgeführt, bei denen Vorsicht geboten ist, zum Beispiel Anwendung bei bestimmten Vorerkrankungen oder Patientengruppen. Es wird auf bestimmte Überwachungspflichten oder bei entsprechenden Medikamenten auf das Potenzial für Missbrauch oder Abhängigkeit eingegangen. Hinweise zur Anwendung bei Patienten mit eingeschränkter Nieren- oder Leberfunktion können ebenfalls enthalten sein.
Damit Patienten die Packungsbeilage und die Beschriftung des Arzneimittels verstehen, müssen die Ergebnisse eines Lesbarkeitstests bei den Behörden vorgelegt werden, um eine Zulassung zu erhalten (AMG § 22).
BfArM: Bundesinstitut für Arzneimittel und Medizinprodukte
CHMP: Committee for Medicinal Products for Human Use, Ausschuss für Humanarzneimittel; Koordinierungsgruppe für gegenseitige Anerkennung und dezentrale Verfahren – Humanarzneimittel
CIOMS: Council for International Organizations of Medical Sciences, Rat für Internationale Organisationen der medizinischen Wissenschaft
CMDh: Coordination Group for Mutual Recognition and Decentralised Procedures
DCP: Decentralised procedure, dezentrales Zulassungsverfahren gleichzeitig in allen gewünschten EU-Mitgliedstaaten
EMA: European Medicines Agency, europäische Zulassungsbehörde
EPAR: European Public Assessment Report, europäischer öffentlicher Bewertungsbericht
FDA: Food and Drug Administration, amerikanische Zulassungsbehörde
MRP: Mutual recognition procedure, gegenseitige Anerkennung; Zulassung eines Arzneimittels in einem Mitgliedstaat der Europäischen Union wird von einem anderen Mitgliedstaat anerkannt.
PRAC: Pharmacovigilance Risk Assessment Committee, Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz
PSUR: Periodic Safety Update Report, regelmäßig aktualisierter Bericht über die Unbedenklichkeit von Arzneimitteln
RMP: Risikomanagementplan


