 |  | Bettina Wick-Urban |
 |
08.02.2026 08:00 Uhr |

Frauen, die potenziell schwanger sein könnten, sind zu Recht besonders kritisch mit der Einnahme von Medikamenten. / © Shutterstock/Grustock
Die Geschichte der Bewertung und Zulassung von Arzneimitteln ist von Tragödien geprägt. Daraus wurden Lehren gezogen, damit den Patienten sichere Medikamente mit dem größtmöglichen Nutzen bei gleichzeitig vertretbarem Risiko zur Verfügung stehen.
Das Jahr 1938 war ein erster Meilenstein, nachdem in den USA nach Einnahme eines Sulfanilamid-Saftes mehr als 100 Patienten starben. Bei der Reformulierung von Tabletten in einen Saft wurde Diethylenglykol verwendet, um den bitteren Geschmack zu überdecken. Der Saft wurde nur auf Geschmack und Geruch getestet, nicht aber auf Sicherheit. Im selben Jahr erließ die amerikanische Regierung ein Gesetz, das die Arzneimittelhersteller dazu verpflichtete, ihre Produkte hinsichtlich der Sicherheit zu testen, und das die Gründung der amerikanischen Zulassungsbehörde FDA (Food and Drug Administration) beschloss (Kasten) (1).

© European Union, 2020
1938, US-Gesetz: Prüfung von Arzneimitteln auf Sicherheit vor der Markteinführung, Gründung der FDA
1962, US-Gesetz: Prüfung von Arzneimitteln auf Wirksamkeit und Sicherheit vor der Markteinführung
1965, erste europäische Pharmagesetzgebung: Prüfung von Arzneimitteln auf Wirksamkeit, Sicherheit und Qualität vor der Markteinführung durch mindestens einen Mitgliedsstaat
1978: Arzneimittelgesetz regelt Zulassung in Deutschland
1998: Verpflichtung der Zulassungsinhaber zur regelmäßigen Bewertung der Unbedenklichkeit ihrer Arzneimittel und strukturierten Nutzen-Risiko-Bewertung nach Markteinführung
2012: Verpflichtung zur Einreichung eines Risikomanagementplans mit dem Zulassungsantrag
2016: Verpflichtung zur Einreichung einer strukturierten Nutzen-Risiko-Bewertung mit dem Zulassungsantrag
Die wohl größte Tragödie, die eine restriktivere Bewertung von Nutzen und Risiko vor der Markteinführung auch in Europa vorantrieb, verursachte Thalidomid.
Das Arzneimittel Contergan® wurde 1957 in Europa als Beruhigungsmittel und später auch bei Angstzuständen, Einschlafstörungen und Morgenübelkeit bei schwangeren Frauen vertrieben, ohne zuvor die Sicherheit bei Einnahme während der Schwangerschaft zu prüfen. Nachdem 1961 Bedenken hinsichtlich toxischer Effekte auf das ungeborene Kind aufkamen, wurde das Medikament im selben Jahr in Europa vom Markt genommen. Aufgrund der Einnahme von Thalidomid während der Schwangerschaft wurden mehr als 10.000 Kinder mit schweren Missbildungen wie Phokomelie (Fehlbildungen der Gliedmaßen) geboren, und es kam zu Tausenden von Fehlgeburten (2).

Viele Menschen, die mehrere Arzneimittel nehmen, wollen sich gut informieren über deren Nebenwirkungen und sonstige Risiken. / © Shutterstock/Kunlathida6242
In Europa wurde daraufhin 1965 die erste Europäische Pharmagesetzgebung eingeführt (3). Diese sah vor, dass ein Arzneimittel nur dann in der damaligen europäischen Wirtschaftsgemeinschaft (EWG) auf den Markt eingeführt werden konnte, wenn die Zulassungsbehörde mindestens eines Mitgliedstaates die Sicherheit, Wirksamkeit und Qualität aufgrund der eingereichten Unterlagen des Arzneimittelherstellers festgestellt hatte (4).
In den USA wurden bereits 1962 die Pharmagesetzgebung verschärft und der Nachweis von Wirksamkeit und Sicherheit verlangt, bevor ein Arzneimittel zugelassen werden konnte (1).
Zu einem weiteren Umdenken bei den Zulassungsbehörden weltweit führten in den 1990er-Jahren die schwerwiegenden Nebenwirkungen nach Markteinführung von Rofecoxib (Vioxx®, vermehrtes Auftreten von Herzinfarkten und Herztod), Cerivastatin (Baycol®, Lipobay®; zum Teil tödlich verlaufende Fälle von Rhabdomyolyse), Terfenadin (Teldane®; schwerwiegende, teils töd-liche Arrhythmien) und Troglitazon (¬Rezulin®; Fälle von Leberversagen). Als »schwerwiegend« werden Nebenwirkungen bezeichnet, die tödlich oder lebensbedrohend sind, eine stationäre Behandlung oder deren Verlängerung erforderlich machen, oder zu bleibender oder schwerwiegender Behinderung, Invalidität, kongenitalen Anomalien oder Geburtsfehlern führen. Diese Nebenwirkungen führten dazu, dass die genannten Arzneimittel vom Markt genommen wurden.
1998 publizierte der Rat für Internationale Organisationen der medizinischen Wissenschaft (CIOMS), der durch die WHO und die Unesco ins Leben gerufen wurde, eine Empfehlung zur systematischen und strukturierten Nutzen-Risiko-Bewertung von auf dem Markt eingeführten Arzneimitteln, die in die Gesetzgebung weltweit Einzug hielt. Diese Empfehlung sollte zu mehr Transparenz beitragen, sodass Risiken frühzeitig entdeckt und bei schwerwiegenden Risiken und negativer Nutzen-Risiko-Bewertung zeitnah Gegenmaßnahmen ergriffen werden können (5).
Der Zulassungsinhaber wurde verpflichtet, regelmäßig die Unbedenklichkeit seines Arzneimittels zu bewerten und das Ergebnis der Analyse inklusive einer strukturierten Nutzen-Risiko-Bewertung bei den Zulassungsbehörden in Form eines PSUR (periodic safety update report; regelmäßiger aktualisierter Unbedenklichkeitsbericht) einzureichen (6). In Deutschland ist diese Verpflichtung im AMG § 63 geregelt.
Mittlerweile werden auch national in Deutschland zugelassene Produkte bei der europäischen Zulassungsbehörde EMA vom Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) bewertet. Der PRAC setzt sich aus Vertretern aus allen Mitgliedstaaten des Europäischen Wirtschaftsraums, wissenschaftlichen Experten sowie Vertretern der Heilberufe und auch der Patientenorganisationen zusammen. Er gibt seine Empfehlung an das Committee for Medicinal Products for Human Use (CHMP, Ausschuss für Humanarzneimittel) für zentral zugelassene Produkte und an das CMDh für dezentral/MRP-zugelassene Produkte. Ergebnisse der PSUR-Bewertung werden auf der EMA-Website und bei national zugelassenen Verfahren auf der BfArM-Website (Bundesinstitut für Arzneimittel und Medizinprodukte) veröffentlicht (7, 8).
Kommt der PRAC zu dem Ergebnis, dass das Nutzen-Risiko-Verhältnis nur dann positiv bleibt, wenn die Fachinformation beziehungsweise die Packungsbeilage geändert werden, ist der Zulassungsinhaber verpflichtet, dies innerhalb eines kurzen Zeitrahmens zu tun (Kasten). Bei sehr schwerwiegenden Risiken kann der Zulassungsinhaber zusätzlich verpflichtet werden, einen Rote-Hand-Brief zu verteilen, um Ärzte und Apotheker schnell zu informieren. Diese sind auch auf der BfArM-Website zu finden (9).

© ABDA
Diclofenac-Präparate (systemische Anwendung)
Azathioprin-Präparate
Um bereits bei der Zulassung eine größtmögliche Transparenz zu schaffen, müssen seit 2016 die Arzneimittelhersteller bereits mit Einreichen des Zulassungsantrags für ein neues Medikament oder bei einer Zulassungserweiterung eine strukturierte und systematische Nutzen-Risiko-Bewertung in der klinischen Zusammenfassung bei den Behörden vorlegen (12, 13). Voraussetzung für die Erteilung der Zulassung ist ein positives Nutzen-Risiko-Verhältnis, das heißt: Der Nutzen des Arzneimittels muss für eine bestimmte Indikation in einer bestimmten Patientengruppe die Risiken überwiegen.
Was beinhaltet die Bewertung? Das Nutzen-Risiko-Verhältnis wird in einer strukturierten wissenschaftlichen Bewertung ermittelt, die quantitative und qualitative Daten integriert. Es gibt verschiedene Ansätze von Zulassungsbehörden und wissenschaftlichen Organisationen, die im Wesentlichen auf drei Säulen beruhen: dem therapeutischen Kontext, den klinisch relevanten Wirksamkeitsparametern und Risiken sowie der Risikooptimierung (Grafik) (13).
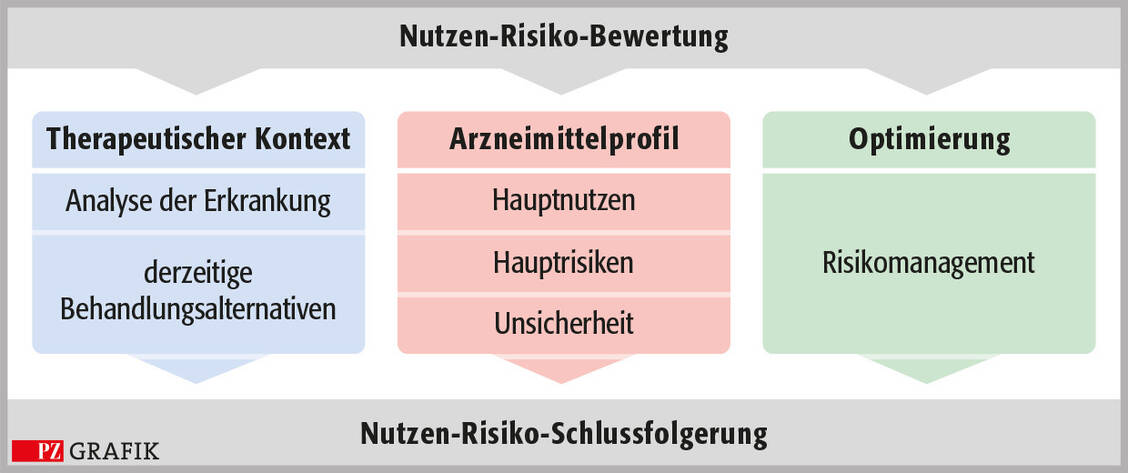
Grafik: Drei Säulen einer strukturierten Nutzen-Risiko-Bewertung; modifiziert nach: Benefit-Risk-Balance for Medicinal Products. CIOMS Working Group report. Geneva, 2025 / © PZ/Stephan Spitzer
Der therapeutische Kontext beschreibt die Erkrankung, deren Symptome, Schweregrad und Häufigkeit sowie – wichtig aus der Perspektive des Patienten – die damit verbundenen Beeinträchtigungen der Lebensqualität, aber auch mögliche Auswirkungen auf das Gesundheitssystem, zum Beispiel erhöhte Kosten durch Operationen oder Krankenhausaufenthalte. Der zweite wichtige Aspekt im therapeutischen Kontext sind die Analyse des Nutzens und der Risiken von alternativen Behandlungsmöglichkeiten sowie die Überlegung, welche Therapielücken aus Sicht der Verschreiber und der Patienten bestehen.
Den Kern der Bewertung stellt die Analyse des Profils des neuen Arzneimittels dar (Grafik, Mitte), das heißt die klinisch relevanten Wirksamkeitsparameter und die positiven Einflüsse auf die gesundheitsbezogene Lebensqualität aus der Perspektive des Patienten sowie andererseits die schwerwiegenden Risiken, die das Nutzen-Risiko-Verhältnis maßgeblich beeinflussen, basierend auf den Daten der (nicht) klinischen Studien. Zum Zeitpunkt der Einreichung des Zulassungsantrags liegen natürlich nur begrenzte Daten vor, zum Beispiel zur Langzeitwirksamkeit, zu seltenen schwerwiegenden Nebenwirkungen oder zu Nebenwirkungen, die erst mit einer gewissen Latenz auftreten, oder zu Patientengruppen, die in die klinischen Studien nicht eingeschlossen wurden. Daher ist der Antragsteller verpflichtet, einen Plan vorzulegen, wie diese Lücken nach der Zulassung geschlossen werden.
Die dritte wichtige Säule der Bewertung ist die Risikooptimierung. Damit ein Arzneimittel ein möglichst günstiges Nutzen-Risiko-Verhältnis hat, muss der Antragsteller die Risikominimierungsmaßnahmen beschreiben. Zu den Routinemaßnahmen gehören zum Beispiel die Verschreibungspflicht, die Packungsgröße oder die Verpackung des Arzneimittels (kindersichere Verschlüsse).
Die wichtigsten Maßnahmen sind in der Fachinformation und der Packungsbeilage festgehalten. Darin wird beschrieben, für welche Erkrankung und welche Patienten ein Produkt in welcher Dosierung zugelassen ist. Weiterhin sind die Kontraindikationen, Wechselwirkungen und Arzneimittelnebenwirkungen beschrieben. Wichtig sind auch die besonderen Warnhinweise und Vorsichtsmaßnahmen für die Anwendung. Hier sind unter anderem schwerwiegende Nebenwirkungen oder Umstände aufgeführt, bei denen Vorsicht geboten ist, zum Beispiel Anwendung bei bestimmten Vorerkrankungen oder Patientengruppen. Es wird auf bestimmte Überwachungspflichten oder bei entsprechenden Medikamenten auf das Potenzial für Missbrauch oder Abhängigkeit eingegangen. Hinweise zur Anwendung bei Patienten mit eingeschränkter Nieren- oder Leberfunktion können ebenfalls enthalten sein.
Damit Patienten die Packungsbeilage und die Beschriftung des Arzneimittels verstehen, müssen die Ergebnisse eines Lesbarkeitstests bei den Behörden vorgelegt werden, um eine Zulassung zu erhalten (AMG § 22).
BfArM: Bundesinstitut für Arzneimittel und Medizinprodukte
CHMP: Committee for Medicinal Products for Human Use, Ausschuss für Humanarzneimittel; Koordinierungsgruppe für gegenseitige Anerkennung und dezentrale Verfahren – Humanarzneimittel
CIOMS: Council for International Organizations of Medical Sciences, Rat für Internationale Organisationen der medizinischen Wissenschaft
CMDh: Coordination Group for Mutual Recognition and Decentralised Procedures
DCP: Decentralised procedure, dezentrales Zulassungsverfahren gleichzeitig in allen gewünschten EU-Mitgliedstaaten
EMA: European Medicines Agency, europäische Zulassungsbehörde
EPAR: European Public Assessment Report, europäischer öffentlicher Bewertungsbericht
FDA: Food and Drug Administration, amerikanische Zulassungsbehörde
MRP: Mutual recognition procedure, gegenseitige Anerkennung; Zulassung eines Arzneimittels in einem Mitgliedstaat der Europäischen Union wird von einem anderen Mitgliedstaat anerkannt.
PRAC: Pharmacovigilance Risk Assessment Committee, Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz
PSUR: Periodic Safety Update Report, regelmäßig aktualisierter Bericht über die Unbedenklichkeit von Arzneimitteln
RMP: Risikomanagementplan
Reichen Routinemaßnahmen nicht aus, um Ärzte, Apotheker und Patienten über die Risiken zu informieren und eine sichere Anwendung des Arzneimittels zu gewährleisten, können mit der Zulassung sogenannte zusätzliche Risikominimierungsmaßnahmen festgelegt werden. In den meisten Fällen handelt es sich um Schulungsmaterial für Ärzte, Apotheker und Patienten wie Leitfäden, Checklisten oder Patientenkarten, die in Deutschland mit der »Blauen Hand« gekennzeichnet sind (behördlich genehmigtes Schulungsmaterial). Auf der Website des BfArM und des Paul-Ehrlich-Instituts sind die aktuellen Schulungsmaterialien für in Deutschland zugelassene Arzneimittel veröffentlicht (14, 15).
Weiterhin kann die Verschreibung oder Abgabe kontrolliert werden, zum Beispiel bei Arzneimitteln mit einem hohen teratogenen Risiko, bei denen auch die Aufklärung der Patientinnen über die Risiken und die Durchführung von Schwangerschaftstests dokumentiert werden muss.

Schulungsmaterial für Ärzte, Apotheker und Patienten, das mit der »Blauen Hand« gekennzeichnet ist, ist behördlich genehmigt und dient der Risikominimierung. / © PZ/D. Hüttemann
Ein Beispiel (14): Für Isotretinoin-haltige Arzneimittel, die bei schweren Formen von Akne eingesetzt werden, darf der Arzt für Frauen nur ein Rezept für den Bedarf von 30 Tagen ausstellen. Dieses kann nur innerhalb von sechs Tagen in der Apotheke eingelöst werden. Der Arzt erhält eine Checkliste mit den Punkten, über die er die Patientin aufklären muss, unter anderem die Risiken, die Verwendung von sicheren Verhütungsmitteln und die monatlichen Schwangerschaftstests. Das Aufklärungsprotokoll muss die Frau unterschreiben. Weiterhin werden die Verhütungsmethode und das Ergebnis des Schwangerschaftstests monatlich dokumentiert.
Für die Apotheke gibt es ebenfalls eine Checkliste. Vor Abgabe muss überprüft werden, dass nicht mehr als der Bedarf von 30 Tagen verordnet wurde und das Rezept nicht älter als sechs Tage ist. Weiterhin sollte das Apothekenteam die Patientin darauf hinweisen, dass sie das Isotretinoin-Arzneimittel nicht mit jemand anderem teilen darf, während der Therapie und einen Monat nach Absetzen nicht Blut spenden darf (da bei einer schwangeren Empfängerin ein Risiko für den Fetus bestehen könnte) und ungenutzte Kapseln bei Therapieende in der Apotheke abgeben soll.
Der Arzt händigt den Patienten eine Patientenkarte aus, in der die Risiken, die Hinweise zur sicheren Kontrazeption und die monatlichen Schwangerschaftstests noch einmal zusammengefasst sind. Männliche und weibliche Patienten werden darauf hingewiesen, dass das Arzneimittel nur für den persönlichen Gebrauch bestimmt ist und sie während der Behandlung und einen Monat danach kein Blut spenden dürfen, da bei einer schwangeren Empfängerin ein Risiko für das ungeborene Kind bestehen würde (14).
Im sogenannten T-Register werden die Verschreibung und Abgabe der teratogenen Lenalidomid-, Pomalidomid- und Thalidomid-haltigen Arzneimittel überwacht (16). Diese Arzneimittel dürfen nur auf Sonderrezepten, sogenannten T-Rezepten, verordnet werden. Vor der ersten Verschreibung muss der Arzt sich beim BfArM registrieren, um die T-Rezeptvordrucke zu bekommen. Ähnlich wie bei Isotretinoin-haltigen Arzneimitteln muss die Aufklärung der Patienten dokumentiert werden.
Die verordnete Menge ist bei gebärfähigen Frauen ebenfalls auf 30 Tage begrenzt und das Rezept muss innerhalb von sechs Tagen eingelöst werden. Die Apotheke muss überprüfen, ob das T-Rezept vollständig und korrekt ausgefüllt und vom registrierten Arzt unterschrieben ist. Den Durchschlag des Rezeptes muss die Apotheke an das BfArM senden.
Für alle Patienten gibt es einen Leitfaden, der über die Risiken aufklärt, sowie eine Patientenkarte, in der bei Frauen auch die Ergebnisse der Schwangerschaftstests dokumentiert werden.
Seit 2012 verlangt das Arzneimittelgesetz (AMG § 4), dass mit einem Antrag auf Zulassung eines Arzneimittels auch ein Risikomanagementplan (RMP) eingereicht werden muss. Der RMP und die darin beschriebenen Maßnahmen zur weiteren Beobachtung und Risikominimierung sind Bestandteil der Zulassung (17).
Zweck des RMP ist es vor allem, zum Zeitpunkt der Zulassung des Arzneimittels bekannte sowie vermutete potenziell wichtige Risiken zu beschreiben und Strategien festzulegen, wie diese in Studien weiter charakterisiert werden können, beziehungsweise Risikominimierungsmaßnahmen für das Arzneimittel festzulegen.
Zusätzlich zu den im RMP aufgeführten Studien wird die Sicherheit von Arzneimitteln nach Marktzulassung routinemäßig über das sogenannte Spontanmeldesystem überwacht.
Im Spontanmeldesystem werden Verdachtsfälle von Nebenwirkungen erfasst, die außerhalb systematischer Untersuchungen »spontan«, zum Beispiel von Patienten während der Einnahme, beobachtet und berichtet werden; dies kann wertvolle Hinweise auf seltene, bislang unbekannte Nebenwirkungen geben. Apotheker sind wie Ärzte aufgrund ihrer Berufsordnungen dazu verpflichtet, Verdachtsfälle unerwünschter Wirkungen zu melden. Aus den Ergebnissen dieser Studien und Meldungen resultieren gegebenenfalls weitere Maßnahmen zur Risikominimierung.

Wenn Patienten über bislang unbekannte Nebenwirkungen eines Arzneimittels berichten, müssen die Heilberufler solche Verdachtsfälle melden. / © Shutterstock/Zamrznuti tonovi
Gibt es neue Erkenntnisse zu den Sicherheitsrisiken eines Arzneimittels oder Änderungen zu Studien oder Risikominimierungsmaßnahmen, muss der RMP überarbeitet und bei den Behörden erneut eingereicht werden. Bei national oder in einem dezentralen oder gegenseitigen Anerkennungsverfahren (DCP/MRP) zugelassenen Produkten findet sich auf der BfArM-Website eine Zusammenfassung des RMP (18).
Bei zentral zugelassenen Produkten werden nach Abschluss des Zulassungsverfahrens der europäische öffentliche Bewertungsreport (EPAR, European Public Assessment Report) sowie der RMP, die Fachinformation und die Packungsbeilage auf der jeweiligen Produktseite der EMA-Website veröffentlicht. Der EPAR fasst die Ergebnisse der klinischen Studien hinsichtlich Nutzen und Risiko zusammen und bewertet, ob das Nutzen-Risiko-Verhältnis für ein Arzneimittel positiv ist und welche Risikominimierungsmaßnahmen festgelegt wurden.
Ein weiteres Sicherheitsnetz in Europa ist die befristete Zulassung eines Arzneimittels zunächst auf fünf Jahre. In diesem Zeitraum steht das Arzneimittel unter zusätzlicher Überwachung, erkennbar an dem schwarzen, auf dem Kopf stehenden Dreieck in der Fachinformation. Die Zulassung wird entfristet, wenn der Zulassungsinhaber eine Nutzen-Risiko-Analyse über den gesamten Zeitraum einreicht und der PRAC im sogenannten Verlängerungsverfahren (Renewal) weiterhin ein positives Nutzen-Risiko-Verhältnis bescheinigt (21).
Leqembi® (Lecanemab) ist ein Arzneimittel zur Behandlung von Erwachsenen mit leichter kognitiver Beeinträchtigung und leichter Demenz in einem frühen Stadium der Alzheimer-Krankheit.
In einer ersten Bewertung der klinischen Studie im Juli 2024 gelangte der CHMP (Ausschuss für Humanarzneimittel der EMA) zu der Auffassung, dass die beobachtete Wirkung von Leqembi hinsichtlich der Verzögerung des kognitiven Abbaus im Vergleich zu Placebo das Risiko schwerwiegender Nebenwirkungen von Amyloid-bedingten Bildgebungsanomalien (ARIA) nicht aufwiegt.
ARIA treten in zwei Formen auf. Bei ARIA-E kommt es hauptsächlich zu Flüssigkeitsansammlungen im Gehirn, bei ARIA-H zu kleinen Blutungen. Obwohl die meisten Fälle von ARIA in der Studie nicht schwerwiegend waren und keine Symptome auslösten, traten bei einigen Patienten schwerwiegende Ereignisse auf. Das ARIA-Risiko war bei Personen mit ApoE ε4-Allel ausgeprägter, insbesondere bei jenen, die zwei Kopien davon hatten.
Daher war der CHMP der Ansicht, dass der Nutzen der Behandlung nicht groß genug sei, um die mit Leqembi verbundenen Risiken aufzuwiegen, und empfahl, die Marktzulassung in der EU zu verweigern.
Bei der erneuten Prüfung im November 2024 schlug der Antragsteller vor, die Anwendung von Leqembi auf Patienten mit nur einer oder keiner Kopie von ApoE4 zu beschränken; dies basierte auf Analysen, bei denen Patienten ausgeschlossen waren, die zwei Kopien des ApoE4-Gens trugen und daher das höchste ARIA-Risiko aufwiesen. Diese Analysen zeigten, dass Patienten mit nur einer oder keiner Kopie von ApoE4 mit einer geringeren Häufigkeit an ARIA-E oder ARIA-H erkrankten als eine breitere Population, während die Wirksamkeit vergleichbar war mit der in der Gesamtpopulation.
In dieser erneuten Prüfung gelangte der CHMP zu dem Schluss, dass der Nutzen von Leqembi bei Patienten mit Alzheimer-Erkrankung im Frühstadium und einer oder keiner Kopie von ApoE4 die Risiken überwiegt, sofern Maßnahmen zur Risikominimierung ergriffen werden, um das Risiko einer schweren und symptomatischen ARIA zu verringern und die Folgen von ARIA langfristig zu überwachen. Die Agentur empfahl daher, die Marktzulassung für Leqembi zu erteilen.
Das Medikament ist über ein kontrolliertes Zugangsprogramm verfügbar, um sicherzustellen, dass es nur bei der empfohlenen Patientengruppe angewendet wird. Vor Behandlungsbeginn sowie vor der 3., 5., 7. und 14. Dosis muss routinemäßig eine MRT-Untersuchung erfolgen, um ARIA zu erkennen, sowie während der Behandlung, wenn Symptome von ARIA auftreten wie Kopfschmerzen, Verwirrtheit, Sehstörungen, Schwindel, Übelkeit und Gehschwierigkeiten.
Ein Leitfaden, eine Checkliste sowie Schulungsprogramme zu ARIA für medizinisches Fachpersonal und eine Patientenkarte sollen über die Risiken aufklären. Darüber hinaus muss der Zulassungsinhaber eine Sicherheitsstudie durchführen, um ARIA-E und ARIA-H weiter zu charakterisieren und die Wirksamkeit der Maßnahmen zur Risikominimierung zu bewerten, sowie eine EU-weite Registerstudie mit Leqembi behandelten Patienten, um die Häufigkeit von Nebenwirkungen einschließlich ARIA abzuschätzen und deren Schweregrad zu bestimmen.
Literatur: 19, 20
Aus den Tragödien in der Vergangenheit wurden Lehren gezogen und strikte Anforderungen an die Marktzulassung und das Verbleiben von Arzneimitteln auf dem Markt festgelegt, damit den Patienten sichere Medikamente mit dem größtmöglichen Nutzen bei vertretbarem Risiko zur Verfügung stehen.
Bei der Zulassung eines Arzneimittels und nach Markteinführung wird regelmäßig das Nutzen-Risiko-Verhältnis geprüft; bei Änderungen werden gegebenenfalls zeitnah Maßnahmen ergriffen. Dieses Verhältnis soll künftig schon in der klinischen Entwicklung bewertet und auch die Perspektive des Patienten noch stärker berücksichtigt werden, um Arzneimittel zu entwickeln, die die Bedürfnisse der Patienten stärker in den Fokus rücken.
Bettina Wick-Urban studierte Pharmazie an der Albert-Ludwigs-Universität in Freiburg. Nach ihrer Promotion in Basel und Freiburg mit einer molekularbiologischen Arbeit im Bereich der experimentellen Onkologie arbeitete sie zunächst als Referentin bei der Arzneimittelinformationsstelle der ABDA. Danach wechselte sie in die pharmazeutische Industrie, wo sie seitdem in verschiedenen Positionen in der klinischen Forschung, Arzneimittelsicherheit und Medizin tätig ist, davon zwei Jahre in den USA. Zwischenzeitlich schloss sie ein Journalismus-Studium ab.


