 |  | Ulrike Holzgrabe |
 |
23.06.2019 08:00 Uhr |

Selektive Estrogen-Rezeptor-Modulatoren haben ihren festen Platz in der Behandlung von Frauen mit Hormonrezeptor-positivem Mammakarzinom. / Foto: Shutterstock/9nong
Schon in den 1930er-Jahren hatten J. W. Cook und E. C. Dodds festgestellt, dass für eine estrogene Wirkung nicht das ganze Estrogen-Molekül notwendig ist. Durch schrittweise strukturelle Veränderung von Anethol (aus Campher) gelangten sie zu dem Dimer Diethylstilbestrol, dessen trans-Isomer dem Estradiol nicht nur strukturell, sondern auch wirkungsmäßig sehr ähnlich war (1).
Mehr als dreißig Jahre später wurden bei den Firmen Merrel sowie ICI zeitgleich die Triarylethylene als lang wirksame Estrogene gefunden, die als Ovulationshemmer designt waren. Mit Clomifen kam allerdings ein Ovulationsauslöser auf den Markt, der sterilen Frauen zur Fertilität verhalf. Das war der Beginn der Karriere der SERM. 1967 kam Clomifen auf den Markt, 1977 Tamoxifen sowie 1996 Toremifen und 1997 Raloxifen (beide zweite Generation). 2013 folgte die dritte Generation mit Ospemifen und Bazedoxifen. Deren vielfältiges Wirkspektrum lässt sich aus dem Wirkmechanismus verstehen, wie im Folgenden erklärt wird.
Im Prinzip wirken Estrogene, ihre Antagonisten und auch SERM durch die Bindung an den Estrogenrezeptor (ER). Dieser ist ein nukleärer Transkriptionsregulator, das heißt er kontrolliert die Expression von Proteinen, die mit der Wirkung von Estradiol in Zusammenhang stehen. Dafür benötigt er fast 300 Proteine! Grundsätzlich besteht der gesamte Aktivierungsprozess aus mehreren Schritten (2), die in Abbildung 1 dargestellt sind.
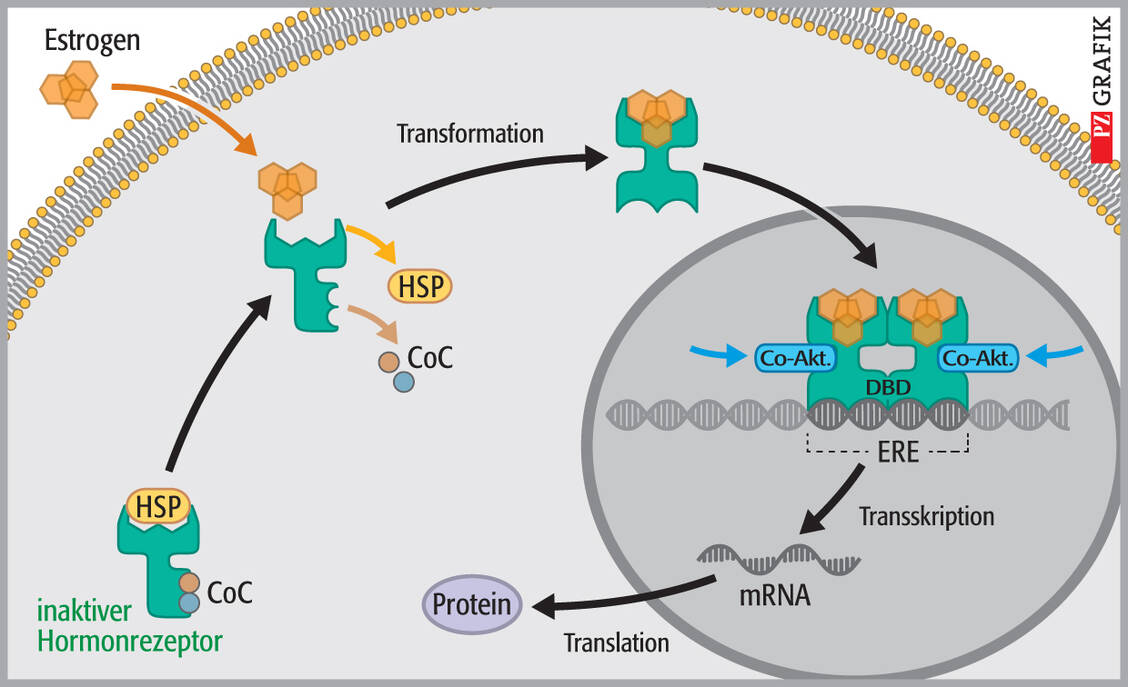
Abbildung 1: Wirkmechanismus der Estrogene und SERM (nach 3)
CoC: Ko-Chaperone, HSP: Heatshock-Proteine, DBD: Bindedomänen des Rezeptorproteins; Co-Akt: Ko-Aktivatoren / Foto: Stephan Spitzer
SERM binden an die gleiche Bindungsstelle am ER wie die Agonisten und können als eine Art Partialagonisten bezeichnet werden, die gewebeabhängig in unterschiedlichem Maß estrogene oder antiestrogene Wirkung induzieren können. Unterschiedliche SERM induzieren unterschiedliche ER-Konformationen und damit auch die Rekrutierung verschiedener Ko-Aktivatoren oder Ko-Repressoren. Dies verleiht jedem SERM sein spezifisches Wirkprofil.
Der Prozess wird weiter verkompliziert, da es nicht nur einen ER gibt, sondern zwei. ER-α findet sich im Uterus, in der Scheide, den Brustdrüsen und Knochen, in den Gefäßen der glatten Muskulatur und im Hypothalamus. ER-β ist am Ovar, an Prostata, Hoden, Lunge, Thymus, Milz und Herz lokalisiert. Das bedeutet, dass die Rezeptorsubtypen eine verschiedene Gewebeverteilung haben.
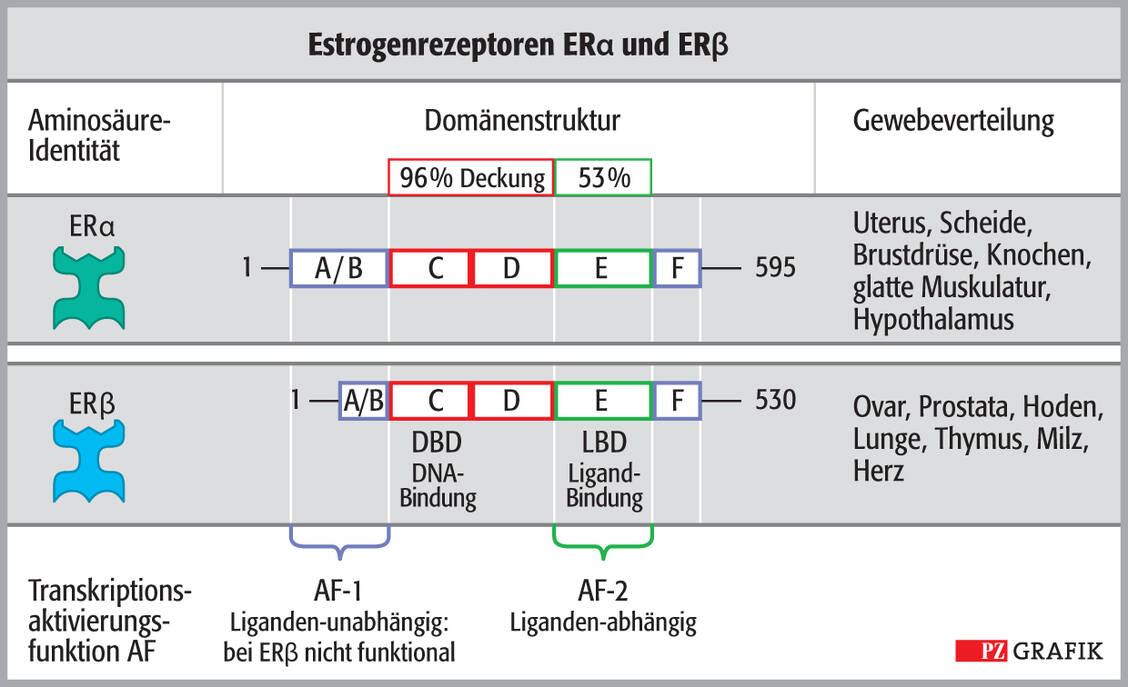
Abbildung 2: Domänenstruktur von ER-α (oben) und ER-β, ihre Aktivationsfaktoren AF-1 und AF-2 sowie ihre Gewebeverteilung; LBD: Ligandenbindestelle, DBD: Bindedomänen des Rezeptorproteins / Foto: Stephan Spitzer
Sie induzieren auch unterschiedliche Effekte: Während ER-α die Genexpression zumeist fördert, hemmt ER-β sie. Zudem verfügen die ER über zwei verschiedene Transkriptionsaktivierungs-Domänen. AF-1 ist konstitutiv aktiv und AF-2 wird Liganden-abhängig aktiviert (Abbildung 2), wobei AF-1 des ER-β aufgrund der verkürzten Aminosäurekette nicht funktional ist. Der endogene Ligand Estradiol wirkt am ER-α agonistisch über AF-1 und AF-2, bei letzterer Domäne Liganden-induziert. Tamoxifen dagegen wirkt über AF-1 agonistisch und über AF-2 antagonistisch.
Die Wirkstoffe der SERM-Gruppe erzeugen ein breites Spektrum an AF-2-Konformationen und stellen damit ein Kontinuum von reinem Agonismus bis zu reinem Antagonismus dar, verbunden mit der Rekrutierung verschiedener Aktivatoren oder Repressoren. Bedenkt man zudem die verschiedene Ausstattung der Zellen mit Ko-Regulatoren, lässt sich verstehen, dass SERM gewebespezifisch die Aktivität der ER ganz unterschiedlich modulieren können.
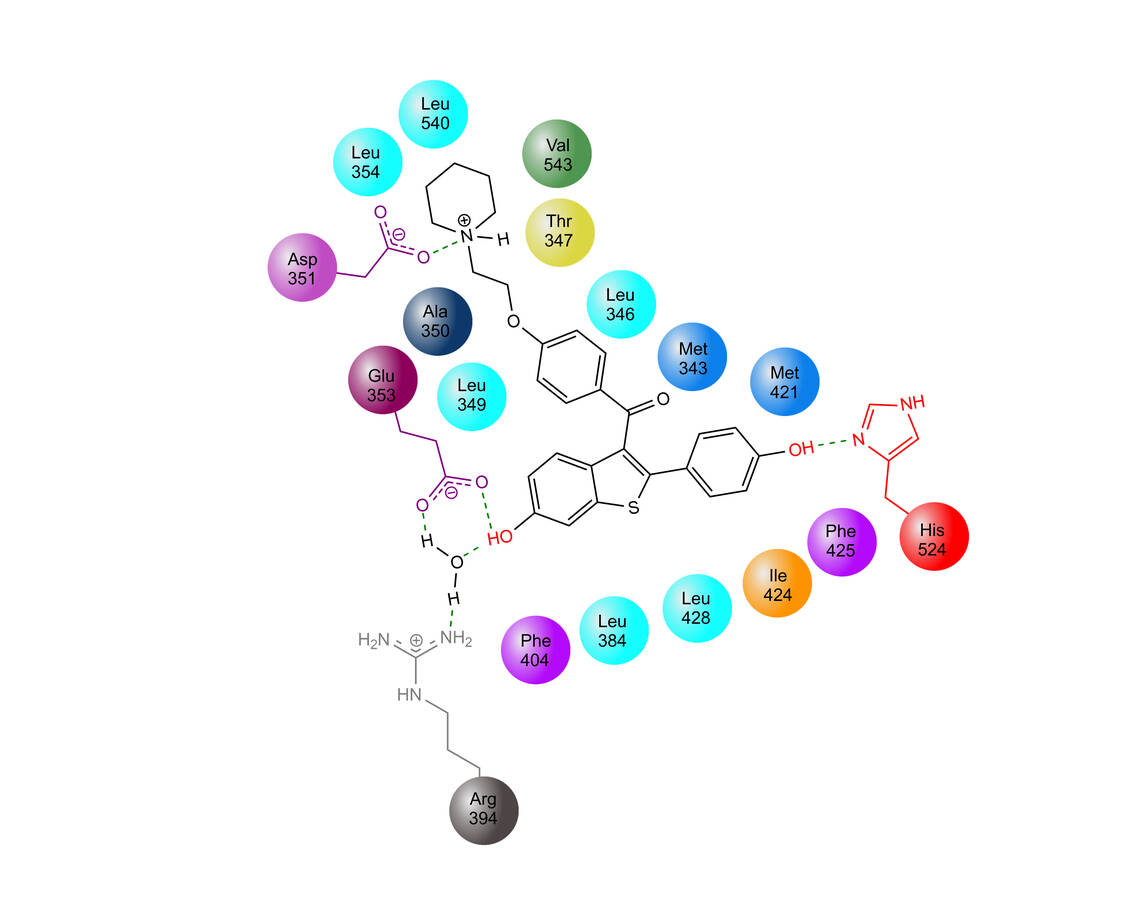
Abbildung 3: Bindemodus von Raloxifen an den Estrogen-Rezeptor (nach 3) / Foto: Mario Wurglics
Erwartungsgemäß binden Estradiol, Hydroxytamoxifen und Raloxifen sehr ähnlich in den ER. Abbildung 3 zeigt dies am Beispiel von Raloxifen.
Die beiden kontralateralen Hydroxylgruppen der Moleküle bilden Wasserstoffbrücken zu einem Imidazolrest des Histidin-524 sowie zu einem Duo aus Arginin-394 und Glutaminsäure-353. Das hydrophobe Grundgerüst wird von aliphatischen Aminosäuren »gehalten«. Die Dimethylammonium-Gruppe des SERM-Ether-Seitenarms geht eine zusätzliche Ion-Ion-Wechselwirkung mit einem Aspartat-350 ein (3, 4) (Abbildung 3). Damit sind die pharmakophoren Elemente und möglichen Strukturvariationen vorgegeben.
Tamoxifen kann man als »First-in-class«-SERM betrachten, das zur Behandlung von Hormon-abhängigem Brustkrebs eingesetzt wird. Schon kurz nach der Markteinführung von Nolvadex® fiel auf, dass nicht alle Patientinnen gleichermaßen von der Therapie profitierten. Dies lässt sich mit dem Metabolismus erklären.
Tamoxifen fehlt die Hydroxylgruppe, die der Phenolstruktur (Ring A) des Estradiols entspricht, und bindet deshalb deutlich schwächer an den ER (1000-fach schwächer als sein Metabolit Hydroxytamoxifen). Tamoxifen ist also ein Prodrug und muss durch Cytochrom-P450-2D6 aktiviert, das heißt hydroxyliert werden. CYP2D6 ist für seinen Polymorphismus bekannt. Eine Reihe von Menschen sind sogenannte »poor metabolizer«, das heißt: Diese Hydroxylierung zu Hydroxytamoxifen und letztlich Endoxifen findet bei ihnen kaum oder nicht statt (Abbildung 4). Daher ist es wichtig, dass vor einer Tamoxifen-Therapie eine Genotypisierung der Patientin erfolgt, um den Therapieerfolg zu ermöglichen (5).
Für die beiden aktiven Tamoxifen-Metaboliten Hydroxytamoxifen und Endoxifen gilt:
Außerdem ist es wichtig für Patienten, die normal gut metabolisieren, keine Komedikation einzunehmen, die CYP2D6 inhibiert. Dazu gehören Antidepressiva wie Paroxetin, Fluoxetin, Bupropion und Duloxetin, aber auch Terfenadin, Ticlopidin und Chinidin.

Frauen, die Tamoxifen einnehmen, sollten pharmazeutisch gut begleitet werden. / Foto: Shutterstock/JPC-Prod
Einig ist man sich darüber, dass eine Komedikation von CYP2D6-Inhibitoren auf jeden Fall vermieden werden sollte. Dagegen ist der Zusammenhang zwischen unwirksamen CYP2D6-Varianten (poor metabolizer), bei denen die Metabolisierung von Tamoxifen und damit der Plasmaspiegel der wirksamen Metaboliten reduziert ist, und Therapieversagen strittig (6).
Zu beachten ist zusätzlich, dass Tamoxifen DNA-reaktive und damit genotoxische Metaboliten bilden kann. Zum einen geschieht dies durch weitere Aromatenhydroxylierung in Nachbarschaft zur ersten Phenolgruppe und Oxidation zum Benzochinon, dessen Michael-System elektrophil an der DNA oder Glutathion angreift. Zum anderen wurde bei Tamoxifen und allen Metaboliten eine ω-1-Oxidation an der Ethylgruppe beobachtet. Die entstandene Hydroxygruppe wird sulfoniert zu einem Sulfat, das als gute Abgangsgruppe nach Eliminierung ein Carbokation hinterlässt, mit dem die DNA alkyliert und damit geschädigt wird. Zur Vermeidung der letztgenannten Reaktion hat man bei Toremifen ein endständiges Chloratom eingeführt.
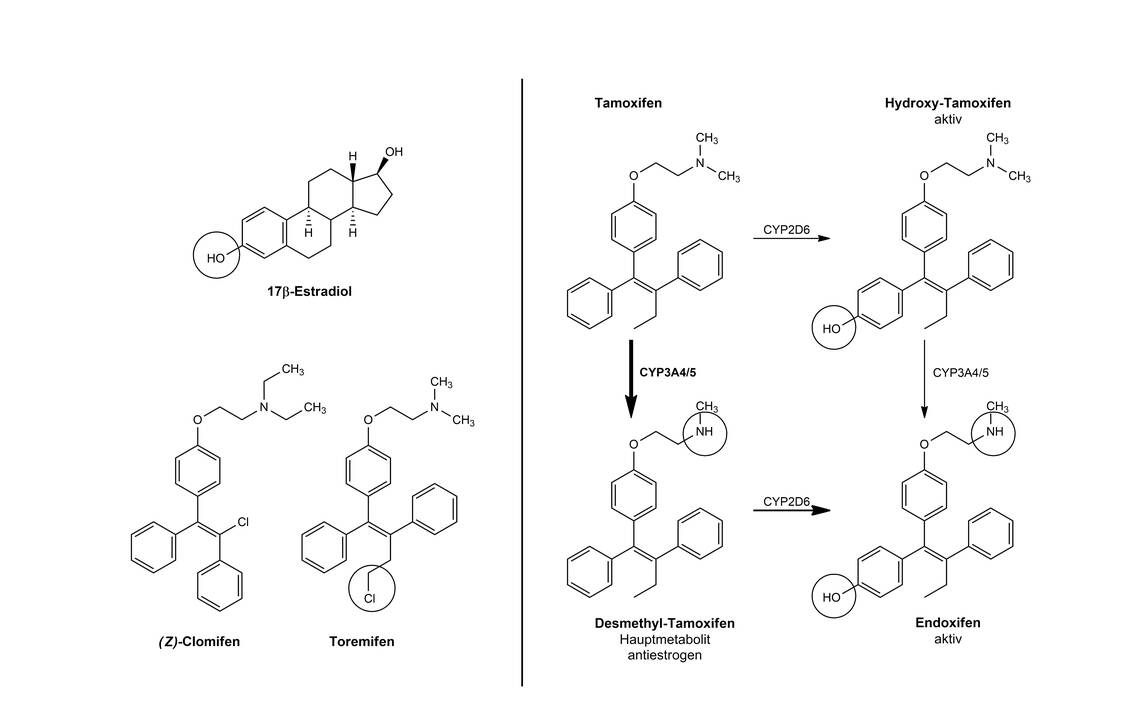
Abbildung 4: Struktureller Vergleich von Estradiol mit Tamoxifen, Clomifen, Toremifen und metabolische Aktivierung von Tamoxifen zu Hydroxytamoxifen und Endoxifen / Foto: Mario Wurglics
Pharmakologisch sind Tamoxifen und seine Metaboliten (Abbildung 4) gemischte Agonisten/Antagonisten an beiden ER. Sie wirken antiestrogen im Brustgewebe und dienen deshalb der adjuvanten Therapie nach Primärbehandlung eines Mammakarzinoms, das als Estrogenrezeptor-positiv diagnostiziert ist. Tamoxifen wird ebenso zur Prophylaxe von Rezidiven bei Hochrisikopatientinnen eingesetzt, allerdings nicht länger als fünf Jahre wegen der dann häufig beobachteten Rückfallgefahr. Aber auch das ist unter Onkologen strittig.
Tamoxifen-Resistenzen sind bei zu langer Gabe häufig. Die Ursachen sind vielfältig. Neben verschiedener Expression von ER-Isoformen, Mutationen in den ER-Rezeptoren, verschiedenen Splice-Varianten, Interferenz bei der Bindung der Ko-Aktivatoren und -Repressoren sowie verschiedener Expression der Ko-regulierenden Proteine (EGF, EGRF, HER2, BRCA1/2) spielt auch der CYP2D6-Polymorphismus eine Rolle (7).
Am Endometrium wirkt Tamoxifen partialagonistisch und kann deshalb zur Hyperplasie, zu Polypen und Tumoren (!) führen. Am Knochen hat Tamoxifen einen estrogenen Effekt und könnte deshalb zur Prophylaxe postmenopausaler Osteoporose eingesetzt werden. Für diese Indikation ist es aber keine erste Wahl.
Die Bioverfügbarkeit von Tamoxifen nach oraler Gabe von 20 mg/d ist nahezu 100 Prozent. Es unterliegt einem minimalen First-Pass-Effekt; die Eliminationshalbwertszeit beträgt sieben Stunden. Nebenwirkungen werden durch den antiestrogenen Effekt regiert: Hitzewallungen, Übelkeit und vaginale Blutungen stehen im Vordergrund.
Neben Tamoxifen spielt in der Therapie des metastasierenden Mammakarzinoms bei postmenopausalen Frauen auch Toremifen eine Rolle (Abbildung 4). Es hat das gleiche pharmakologische Wirkprofil, ist aber weniger potent. Die simple Einführung des Chloratoms an der Ethylgruppe verhindert die metabolische Hydroxylierung an dieser Stelle und damit die Bildung genotoxischer Metaboliten, was einen Vorteil darstellt.
Ospemifen weist statt der Dimethylaminogruppe eine OH-Gruppe auf und kann als Metabolit von Toremifen betrachtet werden. Als Indikation steht nicht die Brustkrebstherapie im Vordergrund, sondern postmenopausale Scheidentrockenheit und symptomatische vulvovaginale Atrophie. Der Gemeinsame Bundesausschuss und das IQWiG sahen allerdings keinen Zusatznutzen einer lokalen Therapie im Vergleich zu einer Estrogenbehandlung (8), was zur Marktrücknahme durch den Hersteller führte. Allerdings laufen weitere Studien für diese Indikation (zum Beispiel (9)).
Die vielleicht größte Bedeutung haben SERM in der Behandlung der Osteoporose. Sie sind zwar alle gemischte Agonisten/Antagonisten und binden am ER-α stärker als an ER-β, wirken aber fast alle estrogen am Knochen. Es sei betont, dass SERM nicht zur Behandlung von klimakterischen Beschwerden zugelassen sind, da sie diese nicht beeinflussen.
Raloxifen, das als SERM der zweiten Generation gilt (Abbildung 5), kann zulassungsgemäß bei Mammakarzinom eingesetzt werden, insbesondere für die adjuvante Therapie bei invasivem Brustkrebs. Prophylaktisch angewendet reduziert es wie Tamoxifen das Brustkrebsrisiko um 50 Prozent. Sein Haupteinsatzgebiet ist allerdings die postmenopausale (!) Osteoporose, und zwar zur Prophylaxe von atraumatischen Wirbelbrüchen, seltener zur Therapie. Achtung: Es hat keine Wirkung bei prämenopausalen Patienten. Es fördert signifikant die Knochenbildung und -dichte und senkt das Bruchrisiko, soll aber weniger wirksam als Alendronat sein.
Raloxifen induziert keine Endometriumhyperplasie und hat keinen Einfluss auf das Risiko von koronaren Herzerkrankungen. Dagegen ist das Risiko für thromboembolische Ereignisse und für Schlaganfall mit tödlicher Folge erhöht (10). Auch wenn die Häufigkeit dieser Nebenwirkungen gering ist, sollte eine Risiko-Nutzen-Analyse angestellt werden. Erwartungsgemäß treten Hitzewallungen und Muskelkrämpfe häufig auf (11).
Raloxifen hat nach oraler Gabe von 60 oder 120 mg/d und guter Resorption (60 Prozent) eine sehr geringe Bioverfügbarkeit (2 Prozent), da es sehr stark präsystemisch am Benzothiophen-Ring und Phenol in der Leber glucuronidiert (mittels UGT1A8/10) wird (Abbildung 5). Durch den enterohepatischen Kreislauf, das heißt Sezernierung über die Galle in den Darm, Dekonjugation im Darm und erneute Resorption, hat es eine Plasmahalbwertszeit von etwa 28 Stunden. Zudem ist es zu 98 Prozent an Protein gebunden.
Die Behandlung der Osteoporose und die Vorbeugung von Frakturen zählen zu den wichtigsten Indikationen der SERM. / Foto: Adobe Stock/ RFBSIP
Bazedoxifen (Conbriza®) lässt sich schon strukturell – Austausch von Benzothiophen- gegen Indolring und Piperidin- gegen Azepinring – als Me-too identifizieren (Abbildung 5). Sowohl das Wirk- wie auch das Nebenwirkungsprofil und die Pharmakokinetik sind nahezu identisch mit Raloxifen (12). Zudem ist es zusammen mit konjugierten Estrogenen (0,45 mg) als Duavive® im Handel, das als Hormonersatztherapie zur Linderung von Hitzewallungen und vulvovaginaler Atrophie eingesetzt werden darf (13). Es ist bei Patientinnen indiziert, die ihre Gebärmutter noch haben, aber mit Progesteron-haltigen Arzneimitteln nicht behandelt werden können.
Lasofoxifen (Fablyn®) wurde ebenso gegen Osteoporose und vulvovaginale Atrophie entwickelt. Auch hier ist die Struktur sehr ähnlich: Die beiden Phenylreste werden durch die Tetralinstruktur in der gleichen Position gehalten wie in der trans-Stellung an der Doppelbindung (Abbildung 5). Trotz allem ist die Affinität von Lasofoxifen zu den ER höher als von Raloxifen, was sich in einer deutlich niedrigeren Dosis (0,25 bis 0,5 mg/d) niederschlägt. Während Wirk- und Nebenwirkungsprofil dem Raloxifen ähneln, ist die Pharmakokinetik von Lasofoxifen viel günstiger, da es kaum präsystemisch glucuronidiert wird. Damit hat es eine gute Bioverfügbarkeit von 62 Prozent.
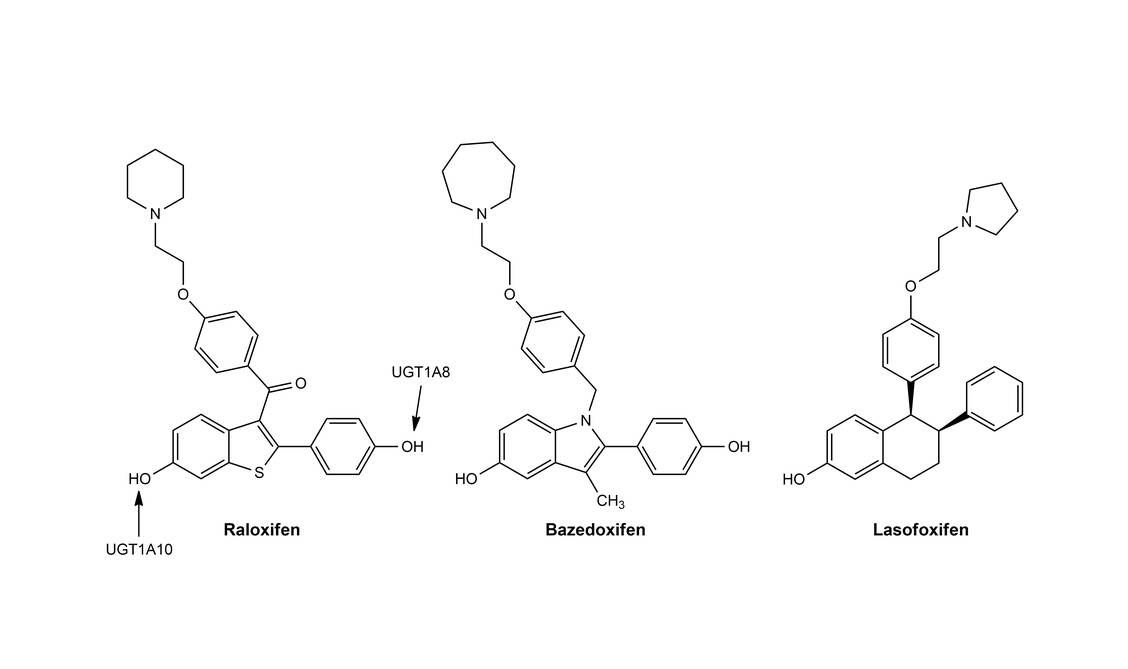
Abbildung 5: Glucuronidierung von Raloxifen (links); Formeln von Bazedoxifen und Lasofoxifen / Foto: Mario Wurglics
Zugelassen ist Lasofoxifen zur Osteoporose-Behandlung im Augenblick nur in Litauen und Portugal. Interessant ist, dass derzeit klinische Phase-III-Studien für die adjuvante Therapie von metastasierendem Brustkrebs laufen.
Last but not least sei in dieser Reihe noch das in Lucknow, Indien, entwickelte Ormeloxifen genannt, in dem das Tetralingerüst gegen Chroman ausgetauscht ist, was zu seinem zweiten Namen Centchroman geführt hat. Dieses nicht-steroidale Kontrazeptivum erzeugt eine Asynchronie zwischen Blastozytenbewegung und endometrischer Empfänglichkeit für die Blastozyten, sodass sich die befruchtete Eizelle im Uterus nicht einnisten kann. Eine Dosis von 30 mg muss nur einmal wöchentlich (t1/2 = 168 h) eingenommen werden (14). Weitere Studien für die Indikationen Brustkrebs und Osteoporose sind in Indien auf dem Weg.
Clomifen, ein SERM der ersten Generation, ist eine Mischung aus cis- (Z = Zuclomifen; Abbildung 4, links) und trans- (E = Enclomifen) Isomeren mit 30 bis 50 Prozent des Z-Diastereomeren. Enclomifen wirkt antiestrogen und Zuclomifen estrogen. Das Z-Isomer hat allerdings nur eine schwache, das heißt partialagonistische Wirkung in Abwesenheit von Estrogen. In der Mischung überwiegt die antiestrogene Komponente von Enclomifens (2).
Vermutlich bindet Clomifen als Antagonist am Steroidrezeptor im Hypothalamus. Damit reagiert die Hypothalamus-Hypophysen-Achse nicht mehr auf das periphere Estrogen, und es findet keine negative Rückkopplung statt. Durch eine gesteigerten Gonadotropin-Ausschüttung kommt es zur Follikelreifung. Die Wirkung ist mit der des Aromatasehemmers Letrozol zu vergleichen (15).
Clomifen hilft Frauen, die an An- oder Oligoovulation leiden, bei der Erfüllung eines Kinderwunsches. Oral werden 50 mg vom fünften bis neunten Zyklustag eingenommen. Häufig reifen allerdings mehrere Follikel, sodass es zu Mehrlingsgeburten kommen kann. 2016 titelte die Süddeutsche Zeitung »Die Zwillingspille« Clomifen, da sich die Zahl der Mehrlingsgeburten verdoppelt hatte.

Clomifen hilft Frauen bei der Erfüllung eines Kinderwunschs. Allerdings kommt es häufig zu Mehrlingsschwangerschaften. / Foto: Adobe Stock/RioPatuca Images
Das E-Isomere Enclomifen (Androxal®) könnte in der Therapie des sekundären Hypogonadismus bei älteren Männern eingesetzt werden, die in der Hypophyse zu wenig Hormon produzieren, um den Hoden zu stimulieren. Dies kann entweder mit Testosteron behandelt werden oder off-label mit Enclomifen, das durch die Blockade des Estrogenrezeptors im Hypothalamus und der erhöhten Gonadotropin-Ausschüttung beim Mann den Hoden stimuliert und die Testosteron-Produktion ankurbelt. Es gibt aber bisher keine Studien, die die Überlegenheit von Enclomifen gegenüber Clomifen belegen (16, 17), und damit auch keine Zulassung, bis weitere Studien die Wirksamkeit eindeutig belegen.
Der Vollständigkeit halber sei noch erwähnt, dass es Klinische Phase-II-Studien gibt für die Anwendung von Zuclomifen zur Bekämpfung von Hitzewellen bei der (antiandrogenen) Prostatakrebstherapie (18).
Clomifen findet auch missbräuchliche Anwendung in der Sportszene. Mit 50 bis 100 mg/d über zwei Wochen eingenommen dient es als Testosteron-Booster zum Muskelaufbau. Deshalb steht es auf der Dopingliste der WADA.
Phytoestrogene wie Isoflavonoide (Genistein, Daidzin), Coumestane, Lignane (Matairesinol, Enterolacton) und Stilbene (Resveratrol) sind schon lange in Diskussion zur Behandlung von Osteoporose, Brustkrebs oder Hitzewallungen. Diese Pflanzeninhaltstoffe sind dem Estradiol und den SERM strukturell sehr ähnlich, was die räumliche Anordnung der Aromaten und der phenolischen OH-Gruppen betrifft.
Von Genistein ist bekannt, dass es in die Bindetasche der Estrogenrezeptoren genauso gut passt wie Estradiol und Raloxifen. Molekuarbiologisch wäre also ein SERM-ähnliches Wirkspektrum zu erwarten. Jedoch gibt es keine klinischen Studien, in denen Phytoestrogene, auch häufig natürliche Phyto-SERM genannt, einen SERM-ähnlichen Effekt beweisen konnten. Die Affinität der Phyto-SERM zu ER-β scheint größer zu sein als zu ER-α, wobei der ER-β die Genexpression eher hemmt als fördert. Hieraus erklärt sich die geringere Potenz der Phyto-SERM und die damit verbundenen höheren Dosen, die für die Induktion eines Effekts notwendig sind.
Trotz vieler widersprüchlicher Ergebnisse aus Tierversuchen fordern viele Mediziner mehr randomisierte Doppelblindstudien mit den Phyto-SERM (19, 20, 21, 22, 23).
Body-Builder, die zum Aufbau ihrer Muskeln zu Testosteron-Derivaten greifen, leiden häufig unter Gynäkomastie, da das überschüssige Testosteron durch CYP19-Enzyme zu Estrogen umgesetzt wird. Hier kann die antiestrogene Wirkung von Clomifen oder Tamoxifen im Brustgewebe helfen, die Gynäkomastie zu unterdrücken. Dafür sind die beiden SERM natürlich nicht zugelassen, aber sie werden im Internet zur »Therapie« der Gynäkomastie gehandelt.
Junge Männer leiden häufig an Pubertätsgynäkomastie, benötigen aber keine Behandlung, da diese physiologisch ist und sich spontan zurückbildet. Hier liegt zeitweise ein Ungleichgewicht zwischen Estrogenen und Testosteron vor.
Kürzlich wurde berichtet, dass Tamoxifen gegen eine ganze Reihe von Bakterien, Parasiten, Pilzen und Viren wirksam sein soll und als Leitstruktur gilt (24). Nach allen Höhen und Tiefen, die diese Arzneistoffgruppe erlebt hat, wäre das der Start einer ganz neuen Karriere.
Literatur bei der Verfasserin


