 |  | Jens Meiler |
| | Clara Schoeder |
 |
08.02.2024 10:30 Uhr |
In den letzten Jahren haben sich die Möglichkeiten zur Anwendung KI-gestützter Methoden zur Wirkstoffentwicklung vervielfältigt. Lange Zeit waren der Zugang zu Daten sowie deren Qualität und Rechenkapazitäten limitierende Faktoren, aber vor allem im strukturbasierten Wirkstoffdesign sind zwei große Errungenschaften zu vermelden:

Ein Mitarbeiter des Instituts für Wirkstoffentwicklung in Leipzig analysiert am Computer Proteinstrukturen. Der linke Bildschirm zeigt eine Struktur im dreidimensionalen Raum. / Foto: IWE/Victoria Most
Beide Methoden zusammengenommen erlauben es heute, fast alle Proteinstrukturen innerhalb von Minuten oder wenigen Stunden vorherzusagen und dann Liganden aus virtuellen Bibliotheken in der Größenordnung von vielen Milliarden Verbindungen im Computer zu testen.
Über viele Jahre hinweg entstanden die ersten Computerprogramme für die Strukturvorhersage von Proteinen und durchliefen eine graduelle qualitative Verbesserung, was durch regelmäßige Überprüfungen in einem gemeinschaftlichen Wettbewerb, dem »Critical Assessment of Techniques for Protein Structure Prediction« (CASP), getestet wurde. Im Jahr 2020 trat »Deep Mind«, eine Google-Tochter, mit »AlphaFold 2« im CASP an und erreichte Vorhersagegenauigkeiten von 80 bis 90 Prozent (7, 8). »AlphaFold 2« ist ein Deep-Learning-Netzwerk, das auf Proteinsequenzen und -Strukturen trainiert worden ist und durch die PDB auf eine große Datenbasis von bereits bekannten Proteinstrukturen zurückgreifen konnte.
Allerdings ist es bei Deep-Learning-Modellen bisher nicht möglich nachzuvollziehen, wie das Modell zu einer Lösung kommt. Daher beschäftigen sich zurzeit viele Wissenschaftler damit, zu verstehen, warum »AlphaFold 2« solch hohe Vorhersagegenauigkeiten erreichen konnte, wie man das Programm weiter verbessern oder es auf andere Anwendungen übertragen kann und was daraus über den Aufbau und das Design von Proteinen abgeleitet werden kann.
Nichtsdestotrotz hat »AlphaFold 2« den Arbeitsalltag in der computergestützten Strukturbiologie drastisch verändert und Prozesse, die früher viele Tage oder Wochen in Anspruch genommen haben, dauern jetzt wenige Stunden.
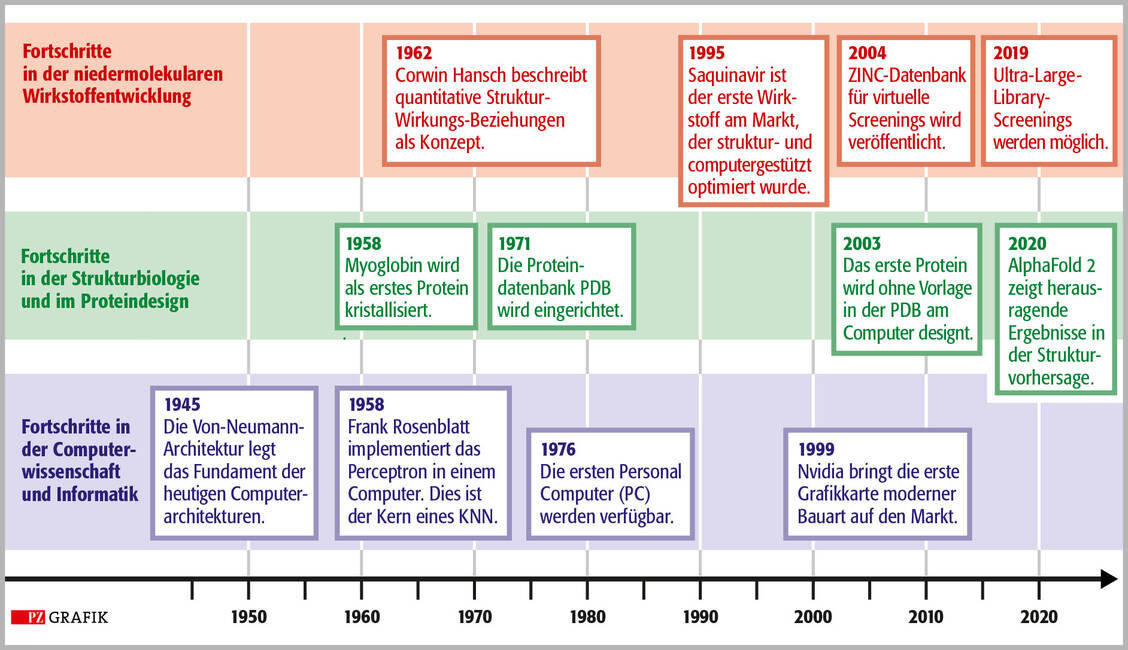
Zeitstrahl mit wichtigen Ereignissen in der Computerentwicklung, der Strukturbiologie und der computergestützten Wirkstoffentwicklung. / Foto: PZ/Stephan Spitzer


