 |  | Jens Meiler |
| | Clara Schoeder |
 |
08.02.2024 10:30 Uhr |

Computergestützte Methoden sind aus der Wirkstoffentwicklung nicht mehr wegzudenken. / Foto: Getty Images/Westend61
Künstliche Intelligenz (KI) ist zurzeit in aller Munde, ob im privaten Umfeld oder der Arbeitswelt. Doch wird selten definiert, was genau darunter eigentlich zu verstehen ist. Tatsächlich existieren verschiedene Definitionen, die bestimmte Methoden des statistischen Lernens ein- oder ausschließen. Oftmals werden alle computergestützten Methoden, die Selbstoptimierung ermöglichen, unter KI zusammengefasst. Dazu zählen zum Beispiel -einfache Verfahren des maschinellen Lernens wie die lineare Regression, aber auch komplexere Methoden wie künstliche neuronale Netze. Die Grundlagen und Konzepte für viele dieser Methoden wurden schon in den 1950er- und 60er-Jahren gelegt, waren aber durch technische Limitationen der Computer und die Unzugänglichkeit zu Daten in ihrer Anwendung begrenzt.
Die ersten Versuche, rationale Wirkstoffentwicklung in Zahlen und Modelle zu überführen, die maschinell verarbeitet und zur Prognose verwendet werden können, stammen aus den 1960er-Jahren. Sie fußen auf den Struktur-Wirkungs-Beziehungen von Arzneistoffen. Medizinisch-chemisch betrachtet, bedeuten diese nichts anderes, als dass die Veränderung einer Molekülstruktur mit einem zu beobachtenden biologischen Effekt in Zusammenhang gebracht wird, zum Beispiel der Bindung an einen Rezeptor oder der Aktivierung eines Signalwegs.
Beim sogenannten ligandenbasierten Wirkstoffdesign werden diese Zusammenhänge in mathematische Modelle überführt. Dies wird durch zwei Faktoren besonders erschwert:
Aus diesen Gründen waren die ersten Modelle zur Optimierung von Wirkstoffen starken Einschränkungen unterworfen und in der Wirkstoffentwicklung nur bedingt anwendbar. Wegweisend waren etwa die Arbeiten des US-amerikanischen Chemikers Corwin Hansch, der quantitative Struktur-Wirkungs-Beziehungen für viele Substanzklassen beschrieben hat (1, 2).

Foto: Getty Images/Hiroshi Watanabe
Künstliche Intelligenz umfasst per Definition der Bundesregierung Methoden zur Lösung von Anwendungsproblemen in der Mathematik und Informatik, die zur Selbstoptimierung fähig sind. Dazu zählen unter anderem Methoden des maschinellen Lernens, aber auch wissensbasierte Systeme.
Maschinelles Lernen bezeichnet die Verwendung von computergestützten Methoden und statistischen Modellen, die Gesetzmäßigkeiten, Abhängigkeiten und Muster aus zugrunde liegenden Daten ableiten. Beispiele für maschinelles Lernen sind künstliche neuronale Netzwerke, Support-Vektor-Maschinen sowie die Anwendung von linearer Regression.
Künstliche oder artifizielle neuronale Netzwerke (KNN oder ANN) wurden nach dem Beispiel des Neurons im Nervensystem entwickelt. Anhand von Testdaten werden Gewichtungen von verschiedenen Parametern bestimmt, wobei das Ergebnis der Zuordnungsvorhersage dazu dient, die Gewichte so lange anzupassen, bis die Zuordnung optimal ist. Ähnlich zu der Verschaltung von Neuronen im menschlichen Gehirn können diese Netzwerke verschiedene nacheinander geschaltete Neuronen – und damit mehrere Schichten – haben.
Deep Learning oder mehrschichtiges Lernen ist eine Unterordnung der maschinellen Lernverfahren. Sie basieren auf KNN, die viele Schichten haben. Dadurch können deutlich mehr Parameter verarbeitet und berechnet werden, es werden aber auch große Datensätze benötigt. Ein Nachteil ist, dass die Optimierung in den Schichten bisher schwer oder gar nicht nachzuvollziehen ist.
Ligandenbasierte Methoden wurden über die Zeit optimiert, um die genannten Einschränkungen zu adressieren. Die Codierung der räumlichen Darstellung von Molekülen im Computer, die Integration von physikochemischen Parametern wie logP-Werten und komplexere Architekturen wie KNN erlauben seit Mitte der 1990er-Jahre die routinemäßige Anwendung in virtuellen Screeningverfahren.
Screeningverfahren, sowohl im Experiment als auch am Computer, identifizieren immer Verbindungen, die mit einer Zielstruktur interagieren oder eine Wirkung auslösen.
Besonders im Zusammenspiel mit Daten aus Hochdurchsatz-Screeningverfahren ist virtuelles Screening attraktiv, da so biologische Aktivitäten für eine hohe Anzahl an Liganden vorliegen. Das Hochdurchsatz-Screening ist ein experimentelles Screeningverfahren, bei dem Verbindungen in biologischen Testverfahren identifiziert werden. Diese Verfahren sind allerdings zeitaufwendig und teuer. In Hochdurchsatz-Screeningverfahren sollen viele Zehntausende Verbindungen möglichst automatisiert und schnell getestet werden. Virtuelles Screening hingegen bezeichnet das Screenen von Verbindungen aus virtuellen Liganden-Bibliotheken.
Virtuelle Screeningverfahren sind dabei vor allem nützlich, um die chemische Diversität der Liganden zu erweitern. Hintergrund hierfür ist, dass obwohl in Hochdurchsatz-Screeningverfahren viele Tausende, Zehntausende oder Hundertausende Verbindungen getestet werden, sie immer nur einen Bruchteil der chemischen Diversität von Arzneistoffen darstellen, die sich auf schätzungsweise 1060 mögliche Verbindungen beläuft (3). Eine solche Anzahl an Verbindungen darzustellen, ist synthetisch gar nicht möglich, unter anderem, weil nicht genug Atome im Universum dafür vorhanden wären.
Klassischerweise beschränkt man virtuelle Screenings nur auf chemische Verbindungen, die auch synthetisierbar sind und wirkstoffartige Eigenschaften besitzen. Solche Verbindungen werden in Datenbanken gespeichert. Als Standard gilt die ZINC-Datenbank (4). Typischerweise wird zur Wirkstoffentwicklung dann eine limitierte Anzahl der virtuell gescreenten Verbindungen bestellt und experimentell auf ihre Aktivität untersucht. Dies limitiert die Zahl zu testender Verbindungen. Oft lassen sich zu diesem Zeitpunkt kritische Strukturelemente ableiten, die für die nachfolgende Optimierung wichtig sind.
Im Gegensatz zu den ligandenbasierten Methoden, die allein den molekularen Aufbau des Wirkstoffmoleküls mit der Aktivität korrelieren, ermöglicht das strukturbasierte Wirkstoffdesign, die Interaktion des Liganden mit der Zielstruktur, also dem Target, in Betracht zu ziehen. Die Zielstruktur wird zur Identifizierung und Optimierung von Wirkstoffen genutzt und muss dazu auf molekularer Ebene aufgeklärt sein. Dies ist erst möglich, seitdem es Verfahren gibt, die Proteinstrukturen auflösen und bildlich darstellen können. Es ist die zentrale Aufgabe der Strukturbiologie, die Funktion von Proteinen aus der Struktur und damit aus der Sequenz abzuleiten.
Myoglobin war 1958 das erste Protein, das durch Röntgenkristallstrukturanalyse aufgeklärt wurde (5). Heutzutage existieren mit biomolekularer Kernspinresonanzspektroskopie (NMR) und der Kryo-Elektronenmikroskopie zwei weitere Methoden zur Aufklärung von Proteinstrukturen, die dazu beigetragen haben, dass Tausende Strukturen in einer öffentlichen Datenbank, der Proteindatenbank (PDB), abgelegt wurden (6).
Beim strukturbasierten Wirkstoffdesign wird im Zuge der Entwicklung idealerweise ein Ligand mit seinem Interaktionspartner im Komplex kristallisiert. Da dies experimentell nicht trivial ist und oft unter hohem Einsatz von Material und Zeit erfolgt, kommt die computergestützte Vorhersage von Ligand-Protein-Interaktionen regelmäßig zum Einsatz. Dabei dockt ein Ligand virtuell in eine Bindungstasche. Anhand dessen können Hypothesen über Struktur-Wirkungs-Beziehungen erstellt werden, die durch Synthese und biologische Testung bestätigt werden können.
In der Vergangenheit war strukturbasiertes Wirkstoffdesign durch die experimentell aufwendige Überprüfung und die Anwesenheit einer Template-Proteinstruktur in der PDB limitiert. Mit der Sequenzierung des humanen Genoms im Jahr 2003 wurde die Strukturvorhersage Kernaufgabe der Bioinformatik. Sie ist der Schlüssel zur allgemeinen Anwendbarkeit von strukturbasiertem Wirkstoffdesign.
Als erster Wirkstoff, der unter Zuhilfenahme computergestützter Methoden entwickelt wurde, gilt Saquinavir. Die HIV-Pandemie in den 1980er- und 1990er-Jahren erforderte eine rasche Arzneistoffentwicklung, die sich aller neuen Methoden bediente und so strukturbasiertes Wirkstoffdesign an der HIV-Protease ermöglichte. Seitdem sind strukturbasierte und computergestützte Methoden aus der Wirkstoffentwicklung nicht mehr wegzudenken.
Im ligandenbasierten Wirkstoffdesign werden Modelle erstellt, um die biologische Aktivität und die chemische Struktur von Liganden miteinander in Korrelation zu bringen. Diese Modelle werden verwendet, um neue Wirkstoffe in virtuellen Bibliotheken zu identifizieren. Die Zielstruktur muss dazu nicht bekannt sein.
Oft sind diese Methoden schnell und brauchen vergleichsweise wenig Computerressourcen. Es wird allerdings eine gewisse Anzahl an aktiven Liganden mit chemischer Diversität benötigt, um robuste Modelle zu generieren. Ligandenbasierte Wirkstoffentwicklung wird vor allem für das schnelle virtuelle Screening herangezogen.
Im strukturbasierten Wirkstoffdesign wird die Information über die Zielstruktur für die Identifizierung und Optimierung des Wirkstoffes genutzt. Die Zielstruktur muss auf molekularer Ebene bekannt und aufgeklärt sein. Dies kann durch Röntgenkristallografie, biomolekulare NMR oder Kryo-Elektronenmikroskopie geschehen. Diese Methoden sind aufwendig, teuer und es gibt keine Garantie auf Erfolg. Daher behilft man sich mit computergestützter Strukturvorhersage, die durch Deep-Learning-Modelle schneller und genauer geworden ist. Wirkstoffe können anschließend in Bindungstaschen platziert, optimiert und designt werden. Traditionell werden strukturbasierte Methoden für die rationale Optimierung von Wirkstoffen verwendet.
In den letzten Jahren haben sich die Möglichkeiten zur Anwendung KI-gestützter Methoden zur Wirkstoffentwicklung vervielfältigt. Lange Zeit waren der Zugang zu Daten sowie deren Qualität und Rechenkapazitäten limitierende Faktoren, aber vor allem im strukturbasierten Wirkstoffdesign sind zwei große Errungenschaften zu vermelden:

Ein Mitarbeiter des Instituts für Wirkstoffentwicklung in Leipzig analysiert am Computer Proteinstrukturen. Der linke Bildschirm zeigt eine Struktur im dreidimensionalen Raum. / Foto: IWE/Victoria Most
Beide Methoden zusammengenommen erlauben es heute, fast alle Proteinstrukturen innerhalb von Minuten oder wenigen Stunden vorherzusagen und dann Liganden aus virtuellen Bibliotheken in der Größenordnung von vielen Milliarden Verbindungen im Computer zu testen.
Über viele Jahre hinweg entstanden die ersten Computerprogramme für die Strukturvorhersage von Proteinen und durchliefen eine graduelle qualitative Verbesserung, was durch regelmäßige Überprüfungen in einem gemeinschaftlichen Wettbewerb, dem »Critical Assessment of Techniques for Protein Structure Prediction« (CASP), getestet wurde. Im Jahr 2020 trat »Deep Mind«, eine Google-Tochter, mit »AlphaFold 2« im CASP an und erreichte Vorhersagegenauigkeiten von 80 bis 90 Prozent (7, 8). »AlphaFold 2« ist ein Deep-Learning-Netzwerk, das auf Proteinsequenzen und -Strukturen trainiert worden ist und durch die PDB auf eine große Datenbasis von bereits bekannten Proteinstrukturen zurückgreifen konnte.
Allerdings ist es bei Deep-Learning-Modellen bisher nicht möglich nachzuvollziehen, wie das Modell zu einer Lösung kommt. Daher beschäftigen sich zurzeit viele Wissenschaftler damit, zu verstehen, warum »AlphaFold 2« solch hohe Vorhersagegenauigkeiten erreichen konnte, wie man das Programm weiter verbessern oder es auf andere Anwendungen übertragen kann und was daraus über den Aufbau und das Design von Proteinen abgeleitet werden kann.
Nichtsdestotrotz hat »AlphaFold 2« den Arbeitsalltag in der computergestützten Strukturbiologie drastisch verändert und Prozesse, die früher viele Tage oder Wochen in Anspruch genommen haben, dauern jetzt wenige Stunden.
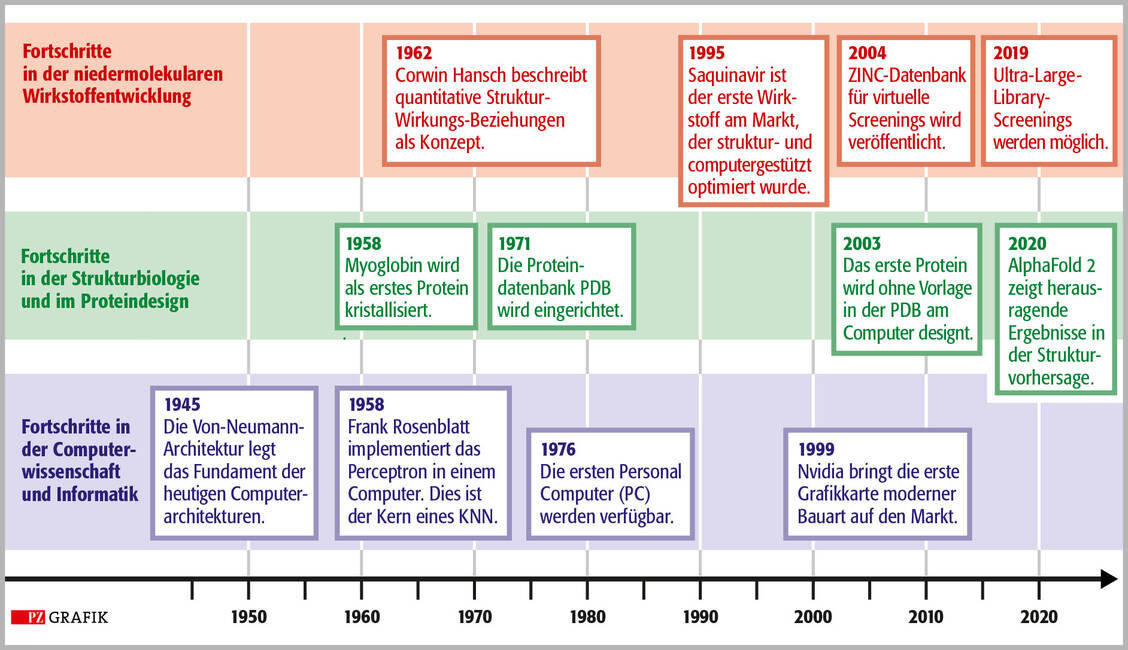
Zeitstrahl mit wichtigen Ereignissen in der Computerentwicklung, der Strukturbiologie und der computergestützten Wirkstoffentwicklung. / Foto: PZ/Stephan Spitzer
Das virtuelle Screening wurde lange mit ligandenbasierten Methoden durchgeführt. Mit der wachsenden Zahl bekannter Proteinstrukturen gewinnt strukturbasiertes Screening aber rapide an Bedeutung.
Es sind Anbieter wie die Firma Enamine Ltd. aus der Ukraine oder Otava Chemicals aus Kanada, die große Bibliotheken chemischer Bausteine bereithalten, aus denen sie via Ein- oder Zweischrittsynthese große Mengen an verschiedenen Verbindungen synthetisieren können. Heute beträgt die Anzahl an Make-on-Demand-Verbindungen der ukrainischen Firma 36 Milliarden mit einer Erfolgsrate von 80 bis 90 Prozent in der Synthese. In dieser Größenordnung spielen experimentelle Screeningverfahren keine Rolle mehr und nur virtuelle Screeningverfahren sind überhaupt in der Lage, solche Datenmengen zu verarbeiten. Es ist daher nicht verwunderlich, dass in der Chemie-Informatik Ultra-Large-Library-Screeningverfahren heutzutage eine große Rolle spielen.
Dabei werden diese riesigen Bibliotheken virtuell an Rezeptoren gedockt und basierend auf den Ergebnissen wenige Hundert Verbindungen ausgewählt, bestellt, synthetisiert und getestet (9, 10). Dieser Prozess gleicht der Suche nach einer Nadel im Heuhaufen. Nur wird der Heuhaufen immer größer.
Erste Studien an G-Protein gekoppelten Rezeptoren wie 5HT2A-Rezeptoren oder D4-Rezeptoren zeigen, dass diese Methoden dazu in der Lage sind, neue Verbindungen zu identifizieren, die bisher unbekannte Grundgerüste haben und mit neuen Eigenschaften aufwarten. Ein Nachteil ist, dass sie auf der Rekombination bekannter Bausteine durch leicht zugängliche Synthesen beruhen und daher keine komplexeren Verbindungen identifizieren können. Auch ist die Anzahl an möglichen Verbindungen inzwischen so hoch, dass es auch großen Computerclustern schwerfällt, alle Verbindungen zu screenen. Methoden des klassischen ligandenbasierten Designs können hier helfen, den Suchraum anhand von Moleküleigenschaften einzugrenzen.
Mit »AlphaFold 2« als Durchbruch in der Strukturvorhersage und Ultra-Large-Library-Screening sind viele Proteine dem strukturbasierten Design zugänglich gemacht worden. In Zukunft wird es vor allem darum gehen, die Methoden zu verfeinern und an spezielle Anforderungen anzupassen.
| Phase der Arzneistoffentwicklung | Anwendungsoptionen für KI |
|---|---|
| Target-Validierung | Expressionsanalysen zum Beispiel aus Krebszellen, Strukturvorhersage |
| Hit-Identifizierung | Virtuelle Screeningverfahren |
| Hit-to-Lead-Optimierung | Strukturbasierte und ligandenbasierte Verfahren zur Wirkstoffoptimierung |
| Präklinische Phasen | Pharmakokinetische Optimierung |
| Klinische Phase I–III | Einschluss- und Ausschluss von Patienten anhand von Metadaten, Korrelation von Wirkung und Nebenwirkung mit Patientendaten, Ursachenforschung für Nebenwirkungen |
| Klinische Phase IV (Pharmakovigilanz) | Korrelation von Wirkung und Nebenwirkung mit Patientendaten, Ursachenforschung für Nebenwirkungen |
Die Natur nutzt nur einen kleinen Teil der möglichen Proteinsequenzen und -strukturen. Im Proteindesign wird eine neue Aminosäuresequenz für eine Proteinstruktur gesucht, die eine bestimmte Funktion erfüllt. Dies wird seit einigen Jahrzehnten computerbasiert versucht, in jüngerer Zeit mit immer mehr Erfolg. Viele proteinogene Wirkstoffe sind von natürlichen Proteinen abgeleitet und modifiziert. Heutzutage beschäftigen sich Forschende vermehrt mit der Frage, ob Proteine komplett neu – ohne natürliche Grundlage – designt werden können. Dann spricht man von De-novo-Design (11).

Am Computer designte Proteine werden im Labor hergestellt und analysiert. Eine Mitarbeiterin des Instituts für Wirkstoffentwicklung in Leipzig bereitet Proben für Gelelektrophoresen vor, um die Größe des jeweiligen Proteins festzustellen. / Foto: IWE/Victoria Most
Mithilfe von Computersoftware, zum Beispiel der Rosetta Software (12, 13), können neue Proteinfaltungen generiert werden, die anschließend sequenzoptimiert werden. Die Rosetta Software basiert auf einem stochastischen Suchverfahren, das eine Energiefunktion, bestehend aus biophysikalischen und wissensbasierten Komponenten, verwendet, um die Ergebnisse zu evaluieren (14).
Das erste Protein, das durch solch ein computergestütztes Verfahren designt wurde, war im Jahr 2003 »Top7«, das damals eine zuvor noch nicht beobachtete Proteinfaltung aufwies (15). Später stellte sich allerdings heraus, dass diese Faltung in der Natur bereits existierte. Die finale Vision des De-novo-Designs ist, dass Proteine und proteinogene Wirkstoffe zielgenau im Computer ohne aufwendige experimentelle Optimierungen entwickelt werden können.
Innerhalb der letzten zwei Jahre wurde eine Vielzahl an Proteindesign-Modellen entwickelt, die KI verwenden, zum Teil mit beeindruckenden Ergebnissen. Dabei gibt es unterschiedliche Ansätze wie große Sprachenmodelle, die Aminosäuresequenzen wie Sprachen interpretieren, strukturbasierte Methoden, die die Wahrscheinlichkeit des Auftretens einer Aminosäure aus dem strukturellen Kontext wiedergeben oder Diffusionsmodelle, die Proteinrückgrate ähnlich wie Bildschärfung aus willkürlich platzierten Atomen und Aminosäuren bauen können.
KI-Methoden wie diese werden zurzeit intensiv erforscht und mit den bisherigen biophysikalischen Methoden verglichen, um ihre Stärken und Grenzen zu evaluieren. Ein großer Vorteil, der sich aus der Anwendung von KI ergibt, ist die Schnelligkeit und einfache Bedienbarkeit des Codes. Es ist aber oft schwer nachzuvollziehen, warum das Modell eine bestimmte Aminosäure wählt.
Durch künstliche Intelligenz könnte die Entwicklung therapeutischer Antikörper künftig kostengünstiger werden. / Foto: Adobe Stock/Dr_Microbe
Zwei sehr wichtige Einsatzgebiete für Proteindesign sind Antikörper und Impfstoffe. Antikörper sind strukturell immer gleich aufgebaut: Zwei leichte und zwei schwere Ketten kommen in der variablen Domäne zu der komplementaritätsbestimmenden Region zusammen, die eine sehr hohe Variabilität aufweist und während der Antikörperreifung optimal an das zu bindende Epitop angepasst wird. Unzählige Methoden sind für das strukturbasierte Design von Antikörpern entworfen worden, um deren Bindung an andere oder mutierte Epitope anzupassen (16). Außerdem wird computergestütztes Design verwendet, um pharmakokinetische Parameter von Antikörpern zu optimieren.
Im Impfstoffdesign werden computergestützte Methoden verwendet, um virale Glykoproteine, die sich auf Virusoberflächen befinden und oft labil sind, zu stabilisieren (17). Diese Methoden sind ein Schwerpunkt der Forschung am Institut für Wirkstoffentwicklung (IWE) in Leipzig, wo sich Wissenschaftlerinnen und Wissenschaftler damit beschäftigen, wie im Pandemiefall am Computer schnell neue Vakzinen entwickelt werden können.
Herausforderungen im Bereich des computergestützten Proteindesigns sind vor allem Fragestellungen, die über den kanonischen Aminosäurecode hinausgehen. Dort ist die Datenlage, um KI-Modelle zu trainieren, nicht gegeben oder lückenhaft. Beispiele sind hier das Design mit nicht-kanonischen Aminosäuren oder post-translationale Modifikationen, die für die chemische Diversifizierung und viele biologische Funktionen essenziell sind.
Im Vergleich zu biologischen Wirkstoffen ist die Entwicklung niedermolekularer Verbindungen, sogenannter Liganden, viel komplexer. Da Proteine meistens aus einer linearen Abfolge von den immer gleichen 20 Aminosäuren bestehen, ist die Komplexität vergleichsweise überschaubar. Bei niedermolekularen Molekülen sind der Verknüpfung von Atomen nur die Grenzen der Chemie gesetzt. So ist es ungleich komplizierter, niedermolekulare Moleküle de novo zu designen. Dies liegt vor allem an der Größe des chemischen Raumes, sprich, der Gesamtzahl an chemischen Molekülen, die als Arzneistoffe einsetzbar wären. Wie beschrieben liegt diese bei 1060.

Niedermolekulare sind im Vergleich zu proteinbasierten Wirkstoffen zwar leichter zu zeichnen, aber deutlich komplexer neu zu designen. / Foto: Getty Images/Westend61
Durch ligandenbasierte Modelle lassen sich chemische Räume strukturieren, sodass zum Beispiel nur nach Vertretern bestimmter Molekülfamilien in Primärscreens gesucht werden kann. Am IWE in Leipzig wird untersucht, wie durch Rekombination von Molekülfragmenten diese gigantischen Suchräume schnell verkleinert werden können. Dabei dienen die möglichen chemischen Reaktionen als Grundlage, man spricht von synthonbasierten Suchverfahren. Am IWE wird mithilfe dieser Methoden Wirkstoffentwicklung an G-Protein gekoppelten Rezeptoren betrieben.
KI wird uns in den kommenden Jahren zunehmend begleiten. In der Wirkstoffentwicklung ergeben sich daraus große Chancen, vor allem in der gezielten Entwicklung von biologischen Wirkstoffen als eine der teuersten Arzneistoffgruppen überhaupt. Der gezielte Einsatz von KI könnte hier eine Kostenreduktion gerade in den frühen Phasen der Entwicklung bedeuten. Dies ist auch eine große Hoffnung für die personalisierte Medizin. Denn für kleine Patientengruppen ist es in wirtschaftlicher Hinsicht schwierig, durch herkömmliche Entwicklungsmethoden neue Wirkstoffe bereitzustellen. Durch KI-gestütztes Design und das experimentelle Nachprüfen nur weniger Wirkstoffkandidaten mit hohen Erfolgsaussichten könnten Kosten gespart werden.
Durch KI-Systeme wie »AlphaFold 2« sind zusätzliche Zielstrukturen für die Wirkstoffentwicklung zugänglich geworden. Wir werden in Zukunft eine Diversifizierung von Zielstrukturen erleben, die strukturbasierten Methoden bisher unzugängliche Proteine und andere Biomoleküle bereitstellt. Die große Herausforderung wird sein, adäquate Testsysteme für das Labor zu generieren. Die Menge an Wirkstoffkandidaten, die experimentell überprüft werden können, wird in Zukunft das Nadelöhr bei der Wirkstoffentwicklung sein.
In der Arzneimittelentwicklung werden computergestützte Methoden immer Hand in Hand mit experimenteller Überprüfung im Rahmen der gesetzlichen Vorgaben gehen. Die Zukunft der Wirkstoffentwicklung könnte allerdings sein, dass eine computergestützte Entwicklung dem Labor vorgeschaltet ist, die die Anzahl an Testkandidaten einengt, bevor es zur experimentellen Testung kommt. Dieser »in-silico-first-Ansatz« wird auch heute zum Teil schon praktiziert.
Eine der größten Herausforderungen für den Einsatz von KI in späteren Phasen der Wirkstoffentwicklung wird die Qualitätskontrolle, Validierung und Verifizierung von Programmen, Softwares und Ergebnissen sein. Wann immer Software unterstützend in klinischen Studien eingesetzt wird, sei es zur Analyse von klinischen Ergebnissen, zur Unterstützung bei der Entscheidungsfindung oder auch zur Diagnose, wird diese Software im Rahmen der »Good Clinical Practice« denselben Qualitätskriterien unterliegen müssen, die für alle anderen verwendeten Methoden gelten. Validierungsprozesse, die für herkömmliche Softwarepakete verfügbar sind, können allerdings zum Teil nicht auf KI-gestützte Anwendungen übertragen werden.
Es ist eine der großen Herausforderungen unserer Zeit, einen klaren gesetzlichen und methodischen Rahmen zu schaffen, um den Einsatz von KI in der Klinik und am Patienten so sicher wie möglich zu gestalten, ohne auf die Vorzüge dieser Technologien zu verzichten.
Der Mensch wird vor allem dann eingreifen müssen, wenn computergestützte Vorhersagen nicht eindeutig sind oder Unsicherheiten aufweisen. Die Beurteilung, Abwägung und risikobasierte Entscheidungen werden für den Menschen als Aufgaben in den Vordergrund rücken. Dies wird auch in der Ausbildung von Wissenschaftlerinnen und Wissenschaftlern beachtet werden müssen.
Jens Meiler hat Chemie an der Universität Leipzig studiert und wurde im Jahr 2001 an der Universität Frankfurt zum Thema biomolekulare NMR promoviert. Darauf folgte ein Aufenthalt in Seattle in den USA an der University of Washington im Labor von David Baker. 2006 wechselte er zur Vanderbilt University in Nashville, Tennessee. Dort führt er bis heute einen Teil seiner Arbeitsgruppe. Seit dem Jahr 2020 ist Meiler Alexander-von-Humboldt-Professor und Professor für pharmazeutische Chemie an der Universität Leipzig. Im Institut für Wirkstoffentwicklung bringt er computergestützte Methodenentwicklung und Translation in experimentelle Validierung zusammen.
Clara T. Schoeder studierte Pharmazie an der Universität Kiel und erhielt 2013 die Approbation als Apothekerin. Zwischen 2013 und 2017 fertigte sie ihre Promotion an der Universität Bonn zum Thema Wirkstoffentwicklung an orphanen G-Protein gekoppelten Rezeptoren und Cannabinoidrezeptoren an. Zwischen 2018 und 2021 war sie an der Vanderbilt University in Nashville in der Arbeitsgruppe von Jens Meiler und James Crowe, um zum Thema computergestütztes Design von Impfstoffen und Antikörpern zu forschen und führt diese Arbeit nun als Gruppenleiterin und seit August 2023 als Juniorprofessorin am Institut für Wirkstoffentwicklung an der Universität Leipzig fort.


