
Weltweit wird an therapeutisch einsetzbaren RNA-Molekülen geforscht. Die Corona-Impfstoffe haben diese Moleküle in der breiten Öffentlichkeit bekannt gemacht. / Foto: Adobe Stock/Evgeniy Kalinovskiy
Die Kürzel RNA oder RNS, Akronyme für »ribonucleic acid« oder »Ribonukleinsäure«, assoziieren die meisten wohl mit messenger RNA (mRNA) als Zwischenspeicher im Rahmen der Decodierung der in der DNA gespeicherten Information, mit transfer RNA (tRNA) als Transportmoleküle für Aminosäuren und Vermittler zwischen mRNA und Protein sowie mit ribosomaler RNA (rRNA) als Strukturelemente und Katalysatoren in den Ribosomen. Dass diese einfach gebauten Biomoleküle mit ihrem Ribose-Phosphat-Rückgrat und den vier Nukleobasen Uracil, Cytosin, Adenin und Guanin auch als Arzneimittel fungieren können, hatte man lange für unmöglich gehalten. Andererseits bietet dieses einfache Bauprinzip über die eindeutigen Komplementaritätsregeln für Nukleinsäuren ein Höchstmaß an Bindegenauigkeit bei einer Wirkstoff-Target-Wechselwirkung.
Jedoch hatte man etliche Vertreter der Molekülspezies RNA in den Zellen lange übersehen beziehungsweise deren Bedeutung und Funktion als »Vorlage« für Arzneistoffe nicht verstanden. Darunter finden sich auch die small interfering RNA (siRNA) – das sind synthetische Analoga der micro RNA (miRNA) –, die als »Sirane« heute eine eigene Gruppe sehr innovativer und potenter Wirkstoffe bilden. Allerdings entpuppen sich die scheinbar einfach gebauten doppelsträngigen RNA-Wirkstoffe bei näherem Hinsehen sowohl strukturell als auch mechanistisch als hochkomplex.
In den Zellen befinden sich jenseits der gut bekannten Vertreter noch viele weitere RNA-Moleküle, darunter die small nuclear RNA (snRNA), die small nucleolar RNA (snoRNA), long non-coding RNA (lncRNA), micro RNA (miRNA), Ribozyme und einige andere mehr. Einen Überblick über die Fülle gibt die Datenbank Rfam (https://rfam.org/), in der man sich beispielsweise alle RNA-Familien anzeigen lassen kann, die im Menschen bisher identifiziert wurden. Diese RNA-Moleküle können vielfältige Funktionen übernehmen: vom Zerschneiden anderer RNA-Moleküle durch Ribozyme bis hin zur Feinmodulierung der Proteinbiosynthese durch miRNA, basierend auf einem Mechanismus, den man als RNA-Interferenz bezeichnet.
Die bahnbrechende Entdeckung der RNA-Interferenz gelang 1998 Andrew Fire und Craig Mello, wofür diese 2006 den Nobelpreis für Physiologie oder Medizin erhielten. Sie zeigten, dass miRNA-Moleküle in der Zelle gebildet werden, um gezielt mRNA stumm zu schalten und dadurch die Biosynthese bestimmter Proteine zu blockieren. Mittlerweile weiß man, dass miRNA lebensnotwendig sind und dass wahrscheinlich 60 bis 90 Prozent unserer Gene auf Ebene der Translation über miRNA reguliert werden (1).
Spätestens seitdem bekannt ist, dass eine Dysregulation bestimmter miRNA kausal an der Entstehung von Tumoren beteiligt ist, rückten diese RNA-Moleküle und der Mechanismus der RNA-Interferenz (RNAi) in den Fokus der Arzneimittelentwicklung (2).
Das Herzstück der RNA-Interferenz bilden kurze doppelsträngige RNA-Moleküle mit einer Länge von 15 bis 30 Basenpaaren. Derartige doppelsträngige RNA-Moleküle können auch als synthetische RNA-Moleküle therapeutisch genutzt werden und werden dann als small interfering RNA (siRNA) bezeichnet.
Wird eine siRNA einem Patienten appliziert und gelingt es ihr, in eine Zelle einzudringen, trifft sie im Zytoplasma auf den sogenannten RNAi-Apparat, den die siRNA benötigen, um »scharf gestellt« zu werden. Dieser RNAi-Apparat wird auch als »RNA-induced silencing complex (RISC)« bezeichnet. Er besteht aus drei Proteinen mit ganz bestimmen Funktionen, die letztlich eine unscheinbare doppelsträngige RNA in ein scharfes Schwert zur Regulation der Genexpression verwandeln (Kasten).
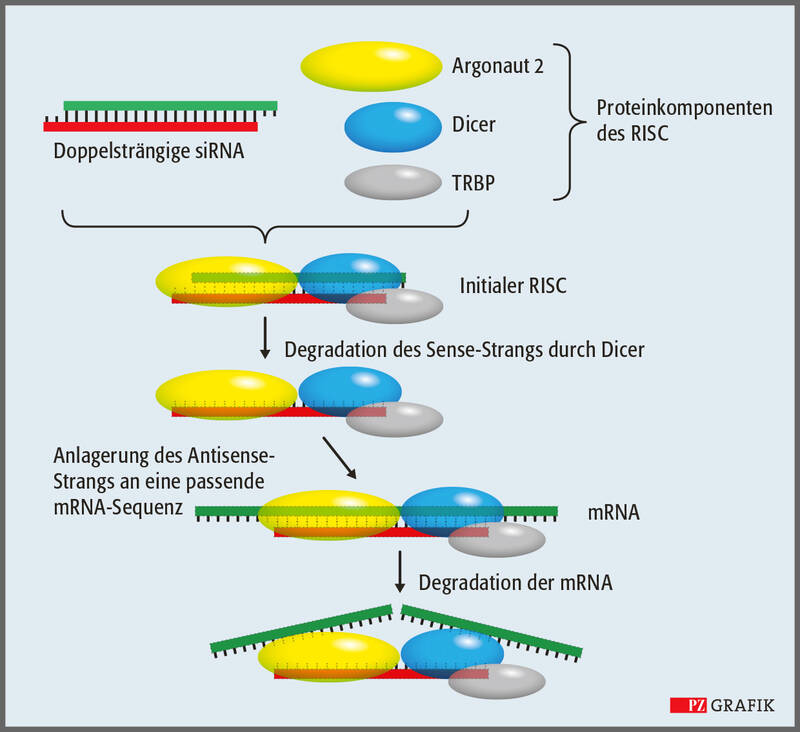
Doppelsträngige siRNA komplexiert zusammen mit den drei Proteinen Argonaut 2, Dicer und TRBBP zum initialen RISC. Danach wird im RISC der Sense-Strang der siRNA abgebaut und der Komplex an eine zum Antisense-Strang komplementäre Sequenz in einer zellulären mRNA dirigiert. Dort wird dann die Ziel-mRNA degradiert. / Foto: PZ/Pfeifer
Ein siRNA-Therapeutikum ist ein Prodrug und wird am RNA-induced silencing complex (RISC) zum eigentlichen Wirkstoff aktiviert. Der RISC besteht aus den Proteinen Dicer, Argonaut 2 (AGO2) und transactivation response element RNA-binding protein (TRBP) (Abbildung).
Dicer trimmt als Endoribonuclease zum einen die RNA-Doppelstränge in die korrekte Länge. Bei diesem Prozess ist auch das RNA-bindende Protein TRBP als eine zentrale Komponente des Dicer-Komplexes beteiligt. Zum anderen baut Dicer den sogenannten »Sense«- oder »Passenger«-Strang ab und legt so mit den komplementären »Antisense«- oder »Guide«-Strang frei, den RISC nun zu derjenigen mRNA lenkt, die über eine passende, zum Guide-Strang komplementäre Sequenz verfügt. Nun kommt AGO2 im RISC zum Einsatz: Dieses Enzym schneidet den mRNA-Strang und baut so die im RISC vorhandene mRNA ab, sodass diese für die Proteinbiosynthese verloren geht. Danach kann der Guide-Strang im RISC an ein weiteres passendes mRNA-Molekül binden und dieses abbauen.
Schätzungen zufolge reichen weniger als 2000 siRNA-Moleküle in einer Zelle aus, um die Übersetzung der Information eines bestimmten Gens in ein Protein komplett stillzulegen – und zwar auf posttranskriptioneller Ebene (3).
Warum ist das siRNA-Konzept als Interventionsoption so interessant? Die meisten etablierten Therapieansätze zielen darauf ab, die Funktion eines bestimmten Proteins zu inhibieren. Das gelingt mal besser und mal schlechter. Man geht davon aus, dass nur ungefähr 1,5 Prozent unseres Genoms für etwa 20.000 Proteine codieren, von denen nur 10 bis 14 Prozent aktive Bindungsstellen haben, die durch kleine Moleküle angesteuert werden können (4, 5). Es bleiben also noch sehr viele Zielstrukturen übrig, die (bisher) nicht durch klassische Wirkstoffe adressiert werden können. Zudem ist es aufwendig und langwierig, ein geeignetes Molekül zu entwickeln, das die gewünschte Wirkung und möglichst wenige Nebenwirkungen hat.
Über siRNA beziehungsweise Sirane könnten theoretisch alle 20.000 Proteine relativ einfach und sehr spezifisch adressiert werden. Der Vorteil ist, dass diese Moleküle ziemlich einfach herstellbar sind – man braucht letztlich nur die richtige Sequenz, die zur zu stoppenden mRNA komplementär ist.
Wie immer, so liegen auch bei Design und Verwendung von siRNA die Probleme in den Details.
Beispielsweise müssen beim Design einer siRNA besondere Anforderungen an die Länge und die Sequenz beachtet werden. Ist die RNA kürzer als 15 Basenpaare, funktioniert sie nicht als RNAi-Molekül. Ist sie länger als 30 Basenpaare, kann sie als vermeintlich virale Infektion interpretiert werden, die die zelluläre Abwehr in Form des angeborenen Immunsystems mobilisiert (6). Außerdem muss es für den RISC eindeutig klar sein, welcher der beiden RNA-Stränge als komplementärer Strang zu einer zellulären RNA fungiert (der Antisense-Strang) und den Abbauprozess der zu blockierenden mRNA steuert. Um dieses Problem zu lösen, werden die Basen möglichst ungleichmäßig verteilt: Der Antisense-Strang der siRNA sollte am 5'-Ende vor allem aus Adenosin- und Uridin-Bausteinen bestehen, während am 3'-Ende Guanosin- und Cytidin-Moleküle vorherrschen sollten (3).
Mittlerweile kennt man etliche Eigenschaften, die eine ideale siRNA haben muss beziehungsweise nicht haben darf, damit die RNA-Interferenz gut funktioniert (Kasten). Diese Optionen lassen sich nicht mehr per Augenschein berücksichtigen, weshalb verschiedene Firmen über Online-Plattformen ein gezieltes Design (mit direkter Bestelloption) für maßgeschneiderte siRNA-Moleküle anbieten.
Doch wie stabilisiert man eine therapeutische RNA, um akzeptable pharmakokinetische Eigenschaften zu erreichen? Denn das ganz große Problem der siRNA sind die Instabilität und Polarität von RNA-Molekülen mit ihren Ribose-Phosphat-Rückgraten. So schätzt man, dass »nackte«, also unmodifizierte und nicht verpackte siRNA-Moleküle im Blut eine Halbwertszeit von gerade einmal fünf Minuten haben (3).
Dieses und andere pharmakokinetische Probleme werden heute durch chemische Modifikationen bestimmter Bausteine innerhalb der siRNA gelöst. Dadurch wird nicht nur sichergestellt, dass die Moleküle nicht mehr so leicht von Nukleasen abgebaut werden, sondern auch, dass die Reaktion des angeborenen Immunsystems auf das fremde Molekül deutlich milder verläuft.
Im Prinzip lassen sich in einer RNA alle drei Grundbausteine – Ribose, Base und Phosphatgruppe – verändern. Allerdings kristallisierten sich gerade die 2'-Modifikation der Ribose und die Verwendung von Phosphorothioaten als besonders geeignet heraus. Ein Blick auf die Struktur der zugelassenen siRNA-Therapeutika zeigt, dass – außer bei Patisiran – alle Nukleotidpositionen chemisch verändert sind (Abbildung 2, Seite 38). Die vermeintlich natürlichen Wirkstoffe sind also »Kunstmoleküle«.
Bleibt noch das Problem der Aufnahme in die Zellen. Aus den Erfahrungen mit zugelassenen Formulierungen, zum Beispiel für die Zytostatika Doxorubicin oder Vincristin, boten sich Lipidnanopartikel (LNP) für die Verpackung der siRNA-Moleküle an. Diese LNP bestehen primär aus Phospholipiden und Cholesterol und ahmen Biomembranen nach. Durch Einlagerung weiterer Bestandteile erhalten die LNP spezielle Eigenschaften. Entsprechend modifizierte LNP können beispielsweise mit dem Apolipoprotein E3 interagieren und werden dadurch zu Hepatozyten transportiert.
Als Alternative zu LNP lassen sich Konjugate herstellen, die aus siRNA und beispielsweise einem Antikörper, Cholesterol oder N-Acetyl-Galactosamin (GalNAc) bestehen. Gerade die Modifikation mit GalNAc hat sich bewährt, um siRNA in Hepatozyten einzuschleusen, da auf der Oberfläche dieser Zellen passende Asialoglycoprotein-Rezeptoren (ASGPR) exprimiert werden. Dazu werden über ein Linkermolekül triantennäre GalNAc-Einheiten kovalent an den Sense-Strang der siRNA gekoppelt. Sehr effizient finden derartige Moleküle nach subkutaner Applikation ihren Weg in die Leber (3).

Foto: Adobe Stock/ipuwadol
Beim Design von RNA-Molekülen, die therapeutisch als Sirane eingesetzt werden sollen, sind zahlreiche Anforderungen zu beachten. Diese betreffen sowohl die siRNA als auch die Ziel-mRNA.
Generelle Eigenschaften von siRNA:
Vermieden werden sollten:
Strukturanforderungen an die Ziel-mRNA:
Aus dem Wirkmechanismus der siRNA-Moleküle lässt sich ableiten, dass all jene Erkrankungen, bei denen ein Protein als »Übeltäter« identifiziert wurde, zumindest in der Theorie über RNA-Interferenz therapiert werden könnten. Denn wenn das Protein nicht mehr (so stark) exprimiert wird, kann dies zur Linderung der Krankheit führen. Bisher sind fünf Sirane für vier Indikationen zugelassen (Tabelle und Abbildung). Alle werden parenteral verabreicht.
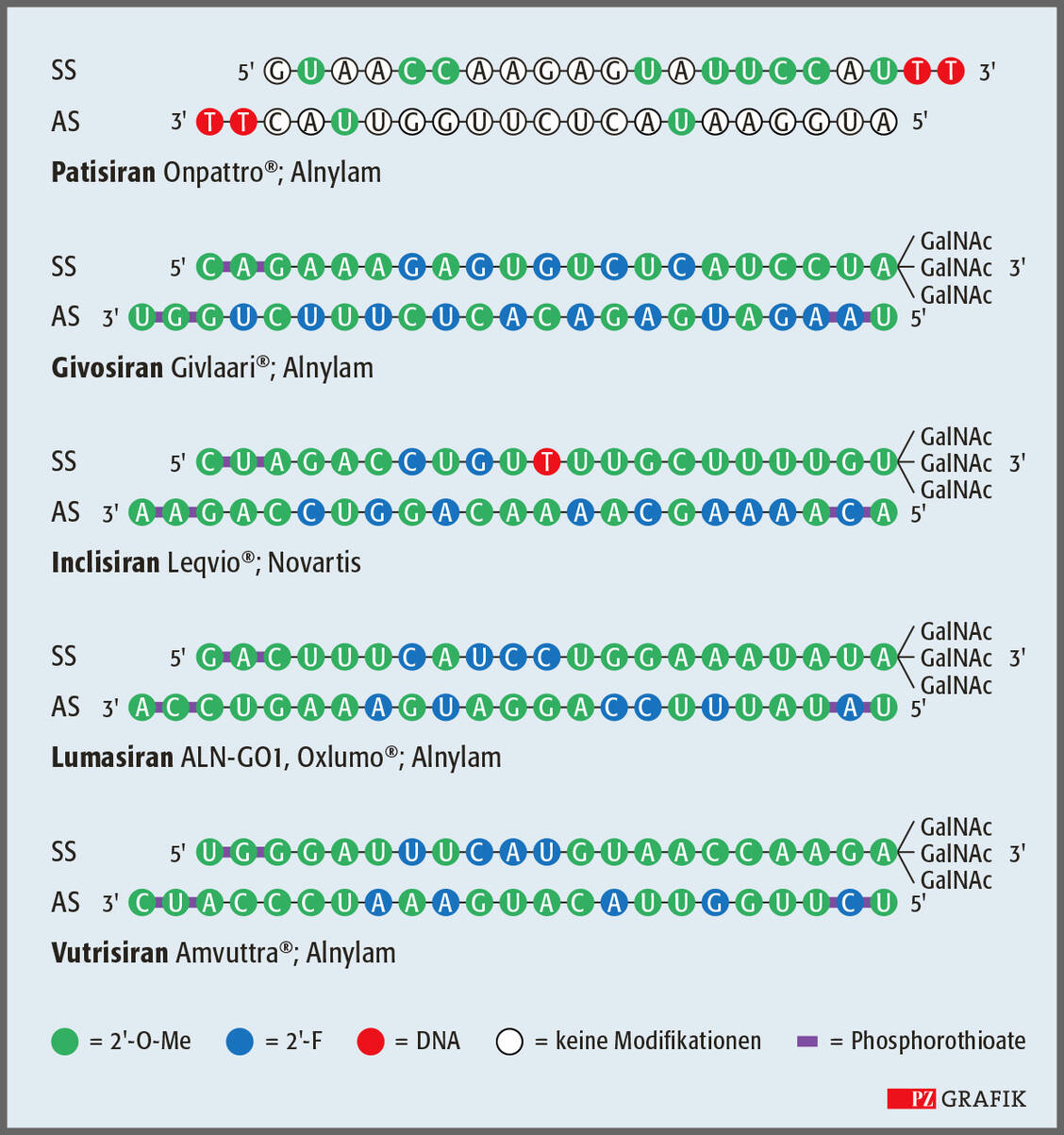
Abbildung: Strukturen der zugelassenen siRNA-Therapeutika. Außer bei Patisiran sind alle Nukleotid-Positionen chemisch verändert (als farbige Kugeln gezeichnet). Meist sind die 2'-Positionen der Ribose durch O-Methylierung (2'-O-Me) oder Fluorierung (2'-F) modifiziert. Zudem werden DNA-Basen (Thymidin) und Phosphorothioate eingebaut. Modif. nach (3). AS: Antisense-Strang; SS: Sense-Strang / Foto: PZ/Pfeifer
Die hereditäre Transthyretin-vermittelte Amyloidose (hATTR) ist eine autosomal dominant vererbte Erkrankung, deren Prävalenz in Europa bei 0,1 pro 10.000 liegt. Es gibt etwa 5000 bis 6000 Patienten. Die physiologische Aufgabe von Transthyretin (TTR) ist der Transport von Thyroxin und Retinol im Körper. Durch eine Mutation im TTR-Gen kommt es zur Bildung von Fibrillen und deren Ablagerung in verschiedenen Geweben. Charakterisiert ist die Erkrankung vor allem durch progressive sensorische und motorische Funktionsstörungen. Unbehandelt sterben die Patienten innerhalb von fünf bis 13 Jahren nach Diagnosestellung.
Die beiden für erwachsene Patienten zugelassenen siRNA-Moleküle Patisiran (Onpattro®), das seit 2018 verfügbar ist, und Vutrisiran (Amvuttra®), das Ende 2022 in den Markt eingeführt wurde, adressieren die TTR-mRNA und reduzieren die Menge an gebildetem Protein um 80 bis 96 Prozent. Damit steht allerdings auch weniger TTR für den Retinol-Transport zur Verfügung, weshalb beim Einsatz beider Arzneimittel eine Supplementation mit etwa 2500 IE Vitamin A pro Tag empfohlen wird.
Ziel aller bisher verfügbaren Sirane ist die Leber. Nur in Hepatozyten kann die siRNA ihre Wirkung entfalten. / Foto: Adobe Stock/natali_mis
Patisiran wird in einer Dosis von 300 µg pro kg Körpergewicht alle drei Wochen intravenös verabreicht. Demgegenüber muss Vutrisiran nur alle drei Monate in einer Dosis von 25 mg subkutan appliziert werden (Tabelle). Dies verdeutlicht die sehr großen pharmakokinetischen Effekte einer konsequenten RNA-Modifikation, wie sie bei Vutrisiran realisiert wurde.
Patisiran erreicht das Leberkompartiment, wo sich die Wirkung in der Hauptsache entfaltet, über Apolipoprotein E3, das sich an die LNP, in die die RNA verpackt ist, anlagert. Apolipoprotein E3 interagiert mit ApoE-bindenden Oberflächenrezeptoren auf Hepatozyten, wodurch dann die LNP in Endosomen der Hepatozyten internalisiert werden. Vutrisiran ist mit N-Acetyl-Galactosamin (GalNAc) gekoppelt.
Pharmakodynamisch unterscheiden sich die beiden Wirkstoffe kaum. Die zeitlich gemittelte prozentuale Reduktion der TTR-Talspiegel bis Monat 18 betrug 84,7 Prozent bei Vutrisiran und 80,6 Prozent bei Patisiran. Die häufigsten Nebenwirkungen unter Patisiran sind periphere Ödeme und infusionsbedingte Reaktionen. Patienten, die Vutrisiran erhalten, klagen gelegentlich über Schmerzen in einer Extremität (15 Prozent) und Arthralgie (11 Prozent).
| Wirkstoff (Arzneimittel, Zulassung) | Erkrankung | Zielprotein | Dosierung/Therapieschema | |||
|---|---|---|---|---|---|---|
| Formulierung als Lipidnanopartikel (DLin-MC3-DMA) | ||||||
| Patisiran | vererbte Transthyretin-vermittelte Amyloidose (hATTR) | Transthyretin (TTR) | 0,3 mg/kg i.v. alle 3 Wochen mit Prämedikation mindestens 60 min vor der Infusion: i.v. Corticosteroid (z.B. 10 mg Dexamethason), 500 mg Paracetamol, i.v.H1- und H2-Blocker (z.B. 50 mg Diphenhydramin und 50 mg Ranitidin) | |||
| Formulierung als N-Acetyl-Galactosamin (GalNAc)-Konjugat | ||||||
| Givosiran (Givlaari®, 2020) | akute hepatische Porphyrie (AHP) | Aminolävulinsäure-Synthase 1 (ALAS-1) | 2,5 mg/kg s.c. einmal monatlich | |||
| Inclisiran (Leqvio®, 2020) | primäre Hypercholesterinämie | Proproteinkonvertase Subtilisin/Kexin Typ 9 (PCSK9) | 284 mg s.c. initial und nach 3 Monaten, danach alle 6 Monate | |||
| Lumasiran (Oxlumo®, 2020) | primäre Hyperoxalurie Typ 1 (PH1) | Glycolatoxidase (GO) | Initialdosis (für 3 Monate):6 mg/kg einmal monatlich (≤ 20 kg KG),3 mg/kg einmal monatlich (> 20 kg)Erhaltungsdosis (beginnend einen Monat nach letzter Initialdosis):3 mg/kg einmal monatlich (< 10 kg),6 mg/kg alle 3 Monate (10 kg bis < 20 kg),3 mg/kg alle 3 Monate (≥ 20 kg) | |||
| Vutrisiran (Amvuttra®, 2022) | Transthyretin-vermittelte Amyloidose (hATTR) | Transthyretin (TTR) | 25 mg s.c. alle 3 Monate |
Mit einer Prävalenz von etwa 1,01 pro 100.000 kommt die seltene Erbkrankheit »Akute hepatische Porphyrie« in Europa vor. Bei dieser Erkrankung ist die Biosynthese des Häms infolge einer Mutation in einem der relevanten Enzyme gestört.
Das geschwindigkeitslimitierende Enzym in der Häm-Biosynthese in der Leber ist die Aminolävulinsäure-Synthase 1 (ALAS-1), deren Expression durch die Konzentration an freiem Häm in der Zelle negativ reguliert wird. Ist eines der nachgeschalteten Biosynthese-Enzyme mutiert und dadurch inaktiv, entsteht nicht ausreichend Häm in der Leberzelle. Infolgedessen wird mehr ALAS-1 gebildet, wodurch wiederum vermehrt die neurotoxischen Substanzen Aminolävulinsäure und Porphobilinogen entstehen. Charakterisiert ist die Erkrankung durch anfallartige, starke abdominale Schmerzen und neurologische Symptome.
Die siRNA Givosiran (Givlaari®) induziert den Abbau der ALAS-1-mRNA und reduziert damit die Blutspiegel der Neurotoxine um etwa 80 bis 90 Prozent. Die Häm-Biosynthese in Erythrozyten ist nicht betroffen, denn hier ist die ALAS-2 aktiv.
Givosiran erhielt 2020 die Zulassung für Patienten ab zwölf Jahren (Tabelle). Da unter der Behandlung die Transaminase-Werte steigen können, sollten vor Beginn der Therapie und während der ersten sechs Behandlungsmonate monatlich Leberfunktionstests erfolgen. Bei klinisch relevanten Transaminase-Erhöhungen ist eine Unterbrechung der Therapie oder ein Abbruch in Betracht zu ziehen. Ebenfalls müssen die Nierenwerte sorgfältig kontrolliert werden, da bei einigen Patienten erhöhte Serumkreatinin- und verringerte eGFR-Werte aufgetreten sind.
Zu bedenken ist auch, dass durch die Behandlung mit Givosiran die Aktivität bestimmter Cytochrom-P-450-Enzyme abnehmen kann. Als Konsequenz des reduzierten hepatischen Metabolismus wurden erhöhte Plasmaspiegel von Coffein (CYP1A2), Dextromethorphan (CYP2D6), Omeprazol (CYP2C19) und Midazolam (CYP3A4) beschrieben. Unter Umständen muss die Dosis einer kritischen Begleitmedikation angepasst werden. Obwohl alle fünf bisher zugelassenen Sirane mit einem Hepatozyten-Targeting ausgestattet sind, tritt das Problem eines reduzierten Metabolismus nur bei Givosiran auf.
Das Target Proproteinkonvertase Subtilisin/Kexin Typ 9 (PCSK9) für eine Therapie der primären Hypercholesterolämie ist schon länger bekannt und wird beispielsweise von den monoklonalen Antikörpern Evolocumab (Repatha®) und Alirocumab (Praluent®) gehemmt. Auf Hepatozyten bindet PCSK9 an LDL-Rezeptoren, woraufhin der Komplex internalisiert und abgebaut wird. Wird die PCSK9 gehemmt, können LDL-Rezeptoren weiterhin LDL-Cholesterol (LDL-C) aus dem Blut abfangen. Alternativ zur Hemmung des Proteins über monoklonale Antikörper kann aber auch die PCSK9-Biosynthese reduziert werden.
Dies gelingt mit Inclisiran (Leqvio®), das seit Anfang 2021 zur Verfügung steht. Dieser siRNA-Wirkstoff induziert über den Abbau der PCSK9-mRNA nach 30 bis 60 Tagen eine mittlere LDL-C-Senkung um 49 bis 51 Prozent. Leqvio® ist zugelassen zur Behandlung von Erwachsenen mit primärer, heterozygot familiärer und nicht familiärer Hypercholesterolämie oder gemischter Dyslipidämie zusätzlich zu einer diätetischen Therapie. Es wird kombiniert mit einem Statin und anderen lipidsenkenden Therapien bei Patienten, die mit der maximal tolerierten Statindosis die LDL-C-Zielwerte nicht erreichen.
Die Primäre Hyperoxalurie Typ 1 (PH1) ist eine seltene, autosomal-rezessiv vererbte Störung des Glyoxylat-Zyklus in Zellen und tritt in Europa mit einer Prävalenz von etwa 0,05 pro 10.000 auf. Ursache für die Störung ist eine mutierte Alaninglyoxylat-Aminotransferase (AGT), wodurch Glyoxylat im Peroxisom nicht mehr in Glycin umgewandelt werden kann. Stattdessen entsteht vermehrt Oxalat im Zytosol, das in Form von Calciumoxalat-Kristallen ausfällt. Die Konsequenzen sind Nierensteine und eine eingeschränkte Nierenfunktion mit möglicher systemischer Oxalose, wodurch Augen, Herzmuskel, Haut und Knochen sowie das ZNS geschädigt werden.
Lumasiran (Oxlumo®) bindet an die mRNA, die für die Glycolatoxidase (GO) codiert. Dieses peroxismale Enzym ist für die Umwandlung von Glycolat in Glyoxylat verantwortlich. Durch die siRNA-Therapie kann die Oxalatmenge im Urin im Vergleich zum Ausgangswert um etwa 65 Prozent reduziert werden.
Das für alle Altersgruppen zugelassene Präparat wird subkutan injiziert, am besten in Unterbauch, Oberarm oder Oberschenkel. Die empfohlene Dosis richtet sich nach dem Körpergewicht und besteht aus Initialdosen einmal monatlich über drei Monate, gefolgt von Erhaltungsdosen (Tabelle, Seite 39). Die Nebenwirkungen sind überschaubar. In den klinischen Studien traten neben Abdominalschmerzen Reaktionen an der Injektionsstelle wie Erytheme, Schmerzen, Juckreiz und Schwellungen auf.
Foto: Adobe Stock/Christoph Burgstedt
Messenger RNA (mRNA):
Mit den Impfstoffen Spikevax® und Comirnaty® gegen COVID-19 erhielten die ersten mRNA-Arzneistoffe die Zulassung. Der Ansatz ist, dass eine bestimmte mRNA in Zellen eingebracht und dort translatiert wird. Dadurch entsteht vorübergehend (transient) das entsprechende Protein, das beispielsweise als Antigen zur Stimulation des Immunsystems dient. Für eine Protein-Ersatztherapie, beispielsweise bei Hämophilie A, ist die Methode eher ungeeignet, weil es zu erheblichen Nebenwirkungen durch Off-target-Effekte kommen kann.
Antisense-Oligonukleotide (ASO):
Einzelsträngige RNA- oder DNA-Oligonukleotide werden in die Zelle eingebracht und binden dort an die zu ihrer eigenen Sequenz komplementäre mRNA (»Sense«-Molekül). Durch diesen doppelsträngigen Bereich kommt es zu einer sterischen Blockade der Funktionalität. Je nachdem, welche Sequenz für das Antisense-Oligonukleotid gewählt wurde und in welcher Region der mRNA die Bindung stattfindet, kommt es zur Inhibition des Translationsstarts, zur Beeinträchtigung des mRNA-Spleißprozesses (Beispiel: Nusinersen in Spinraza®) oder zum Abbau der Ziel-mRNA im DNA-RNA-Komplex durch RNase H (Beispiel: Inotersen in Tegsedi®, Volanesorsen in Waylivra®).
Micro RNA (miRNA):
Kurze, 20 bis 25 Nukleotide lange, doppelsträngige RNA, die natürlicherweise in Zellen vorkommt und dort die Translation bestimmter mRNA steuert. Therapeutisch eingesetzt imitieren die miRNA häufig ihre natürlichen Pendants und stellen deren Funktionalität, beispielsweise in Tumor-zellen, wieder her. Die Wirkweise ähnelt der von siRNA (siehe Haupttext) und führt zur Zerstörung der adressierten mRNA. Im Gegensatz zu siRNA binden miRNA bereits bei einer Teilkomplementarität an die mRNA, vor allem im Bereich der 3'-UTR (nicht translatierte Region). Dadurch kann eine miRNA mehrere unterschiedliche mRNA hemmen.
RNA-Aptamere:
Kurze einzelsträngige RNA-Moleküle, die weniger aufgrund ihrer Sequenz als vielmehr wegen ihrer dreidimensionalen Struktur an unterschiedliche Zielmoleküle binden, zum Beispiel Kohlenhydrate, Proteine oder Nukleinsäuren. Aptamere können dort entweder als Agonisten, Antagonisten oder allgemeine Bindepartner wirken. Das inzwischen wieder vom Markt genommene Pegaptanib (Macugen®) war gegen VEGF gerichtet und zur Therapie der altersabhängigen feuchten Makuladegeneration zugelassen.
Short activating RNA (saRNA):
Kurze, 21 Nukleotide lange, doppelsträngige RNA, von der durch das Protein AGO2 (Argonaut 2) ein Strang (Passenger-Strang) geschnitten wird. Der verbleibende Strang bindet im Komplex mit AGO2 an bestimmte Promotoren von Genen und verstärkt deren Transkription.
Single guide RNA (sgRNA):
Mit dem CRISPR-Cas-System steht ein recht effizientes Werkzeug zur Genomveränderung zur Verfügung. Die Cas-Nuklease benötigt für das zielgenaue Schneiden der DNA in einer Zelle eine sgRNA, die sie an ihre komplementäre Sequenz führt.
Die zugelassenen Sirane zeigen deutlich: Das Prinzip der RNA-Interferenz lässt sich therapeutisch sehr gut nutzen. Voraussetzung ist, dass der Pathomechanismus einer Erkrankung sehr gut aufgeklärt und verstanden ist, denn man muss durch die gezielte Ausschaltung eines Proteins einen Therapieerfolg erzielen können, ohne gleichzeitig einen (lebens)wichtigen biochemischen Prozess zu inhibieren.
Neben Tumorerkrankungen, bei denen beispielsweise onkogene Proteine durch RNA-Interferenz ausgeschaltet werden, können auch wichtige Oberflächenproteine pathogener Viren oder Rezeptoren und Kanalproteine, zum Beispiel bei Augenerkrankungen, adressiert werden – der Fantasie sind praktisch kaum Grenzen gesetzt.
Allerdings stellt die zielgerichtete Applikation immer wieder eine Herausforderung dar. Die bisher zuge-lassenen Sirane steuern alle entweder über Lipidnanopartikel mit Apolipoprotein E3 oder über GalNAc-Konjugate recht erfolgreich Hepatozyten an. Für andere Zielgewebe müssen neue Konjugate gefunden werden. Dies könnten beispielsweise RGD-(Arginin-Glycin-Asparaginsäure-)Motive für die Bindung an Integrine oder Folat für die Interaktion mit Folatrezeptoren auf Tumorzellen sein.
Bleibt noch die Frage nach dem Vehikel. Aus LNP gelangen üblicherweise nur 1 bis 2 Prozent der eingesetzten siRNA aus den Endolysosomen ins Zytoplasma – das sollte noch besser werden. Neben den speziellen Konjugaten und LNP sind auch Exosomen in der Testung. Es gibt wahrscheinlich nicht die eine siRNA-Formulierung, die für alles verwendet werden kann, sondern es muss für jede Indikation die richtige Applikationsform gefunden werden.
Theo Dingermann studierte Pharmazie in Erlangen. Nach Promotion und Habilitation war er bis 2013 Geschäftsführender Direktor des Instituts für Pharmazeutische Biologie an der Goethe-Universität Frankfurt am Main. Jetzt ist er Seniorprofessor der Universität. Die Apotheker kennen ihn als Referenten und Autor von wissenschaftlichen Fach- und Lehrbüchern. Der PZ ist er seit April 2010 als externes Mitglied der Chefredaktion, seit Frühjahr 2019 als einer von drei Chefredakteuren und aktuell als Senior Editor verbunden.
Ilse Zündorf studierte Biologie von 1984 bis 1990 an der Universität Erlangen. Nach einem Forschungsaufenthalt an der Universität von Kentucky, Lexington, USA, wurde sie 1995 am Institut für Pharmazeutische Biologie der Universität Frankfurt promoviert. Zunächst als Akademische Rätin, seit 2001 als Akademische Oberrätin, arbeitet sie am dortigen Institut für Pharmazeutische Biologie. Ihre Forschungsthemen betreffen Herstellung und Charakterisierung monoklonaler Antikörper, Herstellung und Modifikation rekombinanter Antikörperfragmente sowie die Etablierung von zellulären Testsystemen zur Wirkstoffsuche.





