 |  | Annette Rößler |
 |
19.11.2024 18:00 Uhr |

Vor allem Männer ab einem Alter von 60 Jahren können von einer Wildtyp-Transthyretin-Amyloidose betroffen sein. Eine abnehmende körperliche Belastbarkeit zählt neben Gefühlsstörungen und Magen-Darm-Problemen zu den möglichen Symptomen. / © Adobe Stock/SasaStock
Prinzipiell kann eine Amyloidose jedes Organ in Mitleidenschaft ziehen. »Eine Amyloidose ist eine Erkrankung von fehlgefalteten Proteinen, die sich extrazellulär in allen Organen ablagern können«, definierte Dr. Lars Michel von der Klinik für Kardiologie und Angiologie am Westdeutschen Herz- und Gefäßzentrum des Universitätsklinikums Essen kürzlich in einem Vortrag bei den Herztagen der Deutschen Gesellschaft für Kardiologie (DGK). Das Symposium mit Fokus auf die Transthyretin-Amyloidose war von der Firma Alnylam gesponsert.
Es ist nicht immer dasselbe Protein, das durch eine Fehlfaltung die Ursache für eine Amyloidose darstellt. »Es gibt mehr als 35 Proteine, von denen man mittlerweile annimmt, dass sie eine Amyloidose verursachen können. Wahrscheinlich sind es noch viel mehr«, sagte Michel. Für das Herz wichtig seien vor allem zwei Amyloidosen: die Amyloidose aus Transthyretin (ATTR) und die Leichtketten-Amyloidose (AL).
In der Nomenklatur der Amyloidosen steht das A für Amyloidose und die anderen Buchstaben für das fehlgefaltete Protein: TTR für Transthyretin und L für (Immunglobulin-)Leichtkette. Bei der ATTR unterscheidet man weiter in die erbliche (hereditäre) Form (hATTR oder auch vATTR) und den Wildtyp (wtATTR). Die AA oder auch SAA wird durch hohe Konzentrationen von Serum-Amyloid A ausgelöst, die noch selteneren AFib durch mutiertes Fibrinogen und die AApoAI duch mutiertes Apolipoprotein AI. Einen Überblick über die zahlreichen Mutationen, die bei einer Amyloidose relevant sein können, bietet die Internetseite amyloidosismutations.com.
Auf der Internetseite der Initiative Orphanet, die seltene Erkrankungen und Orphan Drugs auflistet, werden die Häufigkeiten wie folgt geschätzt: wtATTR 1 bis 5/10.000 Personen, hATTR 1/450.000 Personen, AL 1/95.800 Personen und AA 1 bis 2/1.000.000 Personen. Die Inzidenz der ATTR steige aber, wie Michel informierte, »weil wir eine immer größere Awareness haben«. Mittlerweile sei bekannt, dass bis zu 16 Prozent der älteren Patienten mit Herzinsuffizienz mit erhaltener Auswurffraktion (HFpEF) oder seniler Aortenklappenstenose eine kardiale ATTR hätten. Bei der großen Mehrheit dieser Patienten liegt eine wtATTR vor.

Die TTR-Fibrillen lagern sich bei einer Amyloidose nicht nur im Herzen ab, sondern auch in anderen Geweben und können etwa ein Karpaltunnelsyndrom verursachen. / © Getty Images/manusapon kasosod
Das Serum-Transportprotein TTR ist ein homotetramerisches Protein; es besteht also aus vier identischen Monomeren. Diese werden zum überwiegenden Teil in der Leber gebildet. Bei Patienten mit ATTR kommt es zum Zerfall des TTR in die Monomere – bei hATTR getriggert durch eine instabile Konformation des TTR und bei wtATTR durch einen bislang unbekannten Auslöser (das TTR ist bei diesen Patienten nicht mutiert). Die Monomere lagern sich dann zu unlöslichen Fibrillen zusammen, vor allem im Herzen, aber auch im Weichteilgewebe.
Eine AL entsteht dagegen durch Störungen in Plasmazellen, in deren Folge diese übermäßige Mengen abnormer Antikörperproteine bilden, die als leichte Ketten bezeichnet werden. Diese bilden unlösliche Fibrillen im Herzen, aber auch in der Haut, den Nieren, den Nerven, den Blutgefäßen, der Zunge, dem Darm, der Leber und der Milz. Behandelt wird eine AL mittels Immun-Chemotherapie.
Die AA wird durch hohe Konzentrationen von Serum-Amyloid A ausgelöst, manifestiert sich vor allem in der Niere und stellt eine mögliche Komplikation von chronisch-entzündlichen oder infektiösen Erkrankungen dar. Die Behandlung besteht in der Therapie der zugrunde liegenden Erkrankung.
Von der wtATTR sind überwiegend Männer jenseits des 60. Lebensjahres betroffen; sie wurde früher auch als senile systemische Amyloidose bezeichnet. In einer alternden Bevölkerung sind daher weiter steigende Fallzahlen zu erwarten. Eine wtATTR gehe mit einer erheblichen Morbidität und Mortalität einher, so Michel. Studien zufolge beträgt die mittlere Lebenserwartung nach der Diagnosestellung 3,5 bis 6 Jahre. Oft werde die Diagnose erst spät gestellt.
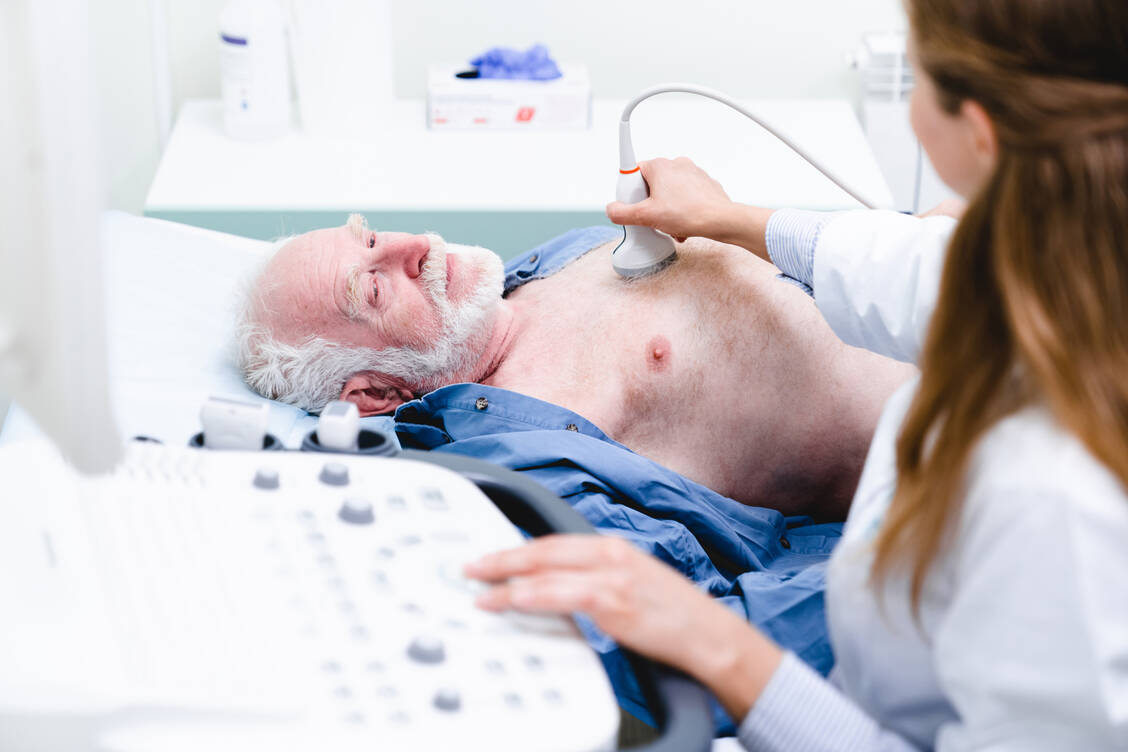
Im Ultraschall sieht man bei Patienten mit kardialer Amyloidose beidseits eine Hypertrophie der Ventrikel. / © Adobe Stock/InsideCreativeHouse
Typisch für eine kardiale Amyloidose sei eine biventrikuläre Hypertrophie, die der Arzt mittels Echokardiografie feststellen könne. »An eine Amyloidose sollte man denken, wenn zwei Kriterien erfüllt sind: eine linksventrikuläre Wanddicke von 12 mm oder mehr plus eine oder mehrere red flags«, so Michel. Zu Letzteren zählten neben kardialen Problemen wie Herzinsuffizienz und Vorhofflimmern etwa ein beidseitiges Karpaltunnelsyndrom, eine Ruptur der distalen Bizepssehne und eine lumbale Spinalkanalstenose (eher typisch für ATTR) oder auch eine Makroglossie (große Zunge, eher typisch für AL).
Auf die Therapieoptionen bei ATTR ging Privatdozentin Dr. Caroline Morbach vom Universitätsklinikum Würzburg ein. Derzeit sei Tafamidis (Vyndaqel®) das einzige zugelassene Medikament für die Behandlung von Patienten mit wtATTR. »Tafamidis ist ein selektiver Stabilisator des TTR-Tetramers«, informierte Morbach. In der Zulassungsstudie ATTR-ACT sei Tafamidis gegenüber Placebo im untersuchten Kollektiv aus Patienten mit wtATTR und hATTR hinsichtlich der Gesamtmortalität, den Hospitalisierungen aufgrund kardiovaskulärer (CV) Ursache, der Abnahme der Sechs-Minuten-Gehstrecke und der Lebensqualität signifikant überlegen gewesen (»New England Journal of Medicine«, NEJM, 2018, DOI: 10.1056/NEJMoa1805689). Die Vorteile durch die Medikation zeigten sich allerdings erst ab einer Therapiedauer von 18 Monaten.
Mit Acoramidis stehe ein weiterer TTR-Stabilisator kurz vor der Zulassung; Anfang des Jahres seien positive Daten der Phase-III-Studie ATTRibute-CM im NEJM publiziert worden (DOI: 10.1056/NEJMoa2305434). »Es gibt eine seltene TTR-Mutation, die eine stärkere Stabilisierung des Tetramers bewirkt. Acoramidis wurde designt, um diese Mutation zu imitieren«, sagte Morbach. Die Gabe des Arzneistoffs führe zu einer nahezu kompletten Stabilisierung der TTR-Tetramere.
In der ATTRibute-CM-Studie sei der Anteil der Patienten mit wtATTR mit 90 Prozent deutlich höher gewesen als in der ATTR-ACT-Studie (76 Prozent). Ein signifikanter Vorteil gegenüber Placebo zeigte sich im hierarchischen Endpunkt, der aus Tod jeglicher Ursache, CV-Hospitalisierung, Veränderung des NT-proBNP und Veränderung der Sechs-Minuten-Gehstrecke zusammengesetzt war. »Auch hier brauchte es eine gewisse Zeit, bis erste Therapieeffekte sichtbar wurden, nämlich 19 bis 24 Monate«, berichtete die Expertin.
Pathophysiologisch eine Stufe früher setzen Medikamente an, die die Produktion des TTR in der Leber unterbinden. In der EU zugelassen sind Patisiran (Onpattro®) und Vutrisiran (Amvuttra®), die auf dem Prinzip der RNA-Interferenz (RNAi) basieren, sowie das Antisense-Oligonukleotid Inotersen (Tegsedi®). Eplontersen (Wainzua®), ein weiteres Antisense-Oligonukleotid, ist von der Europäischen Arzneimittelagentur (EMA) zur Zulassung empfohlen, aber noch nicht zugelassen.
Alle vier genannten Wirkstoffe sind ausschließlich für die Therapie von Patienten mit hATTR und Polyneuropathie bestimmt – noch. Denn die viel größere Indikation kardiale wtATTR befindet sich im Visier zumindest von Patisiran- und Vutrisiran-Hersteller Alnylam. Vutrisiran, das eine Weiterentwicklung von Patisiran darstellt, wurde in der kürzlich publizierten HELIOS-B-Studie erfolgreich auch bei Patienten mit wtATTR getestet (NEJM, DOI: 10.1056/NEJMoa2409134). In dieser Studie machten wtATTR-Patienten einen Anteil von 88 Prozent aus.
Noch ziemlich weit von einer möglichen Zulassung entfernt ist das CRISPR/Cas-basierte Gentherapeutikum NTLA-2001. »Die einmalige intravenöse Applikation von NTLA-2001 bewirkt eine Frameshift-Mutation im TTR-Gen und in der Folge die Drosselung der TTR-Produktion«, erklärte Morbach. Besonders an diesem Ansatz sei, dass hier nicht wie sonst bei der Gentherapie üblich ein viraler Vektor verwendet werde, sondern Lipid-Nanopartikel, die über den LDL-Rezeptor in die Leberzellen eingebracht würden. NTLA-2001 wirke somit spezifisch nur in der Leber. Erste Ergebnisse einer Phase-I-Studie seien »relativ vielversprechend« gewesen (NEJM 2021, DOI: 10.1056/NEJMoa2107454).
Auch Wirkstoffe, die abgelagertes TTR-Amyloid wieder abbauen, gibt es bereits. Der Referentin zufolge handelt es sich um humanisierte monoklonale Antikörper, die an das fehlgefaltete TTR binden. Dabei würden Epitope angesteuert, »die nur in der Fehlfaltung freiliegen und nicht am normal gefalteten Transthyretin«. Die Antikörper binden sowohl an den Monomeren als auch an den abgelagerten Fibrillen. »Durch die Antikörper-Bindung wird die Phagozytose induziert und es kommt zum Abbau von Amyloid-Fibrillen«, sagte Morbach.
Zu einem dieser sogenannten Depleter, NI006, seien bereits Phase-I-Daten publiziert worden (NEJM 2023, DOI: 10.1056/NEJMoa2303765) und eine Phase-III-Studie laufe. Ein weiterer Depleter, NNC6019-0001, werde derzeit in einer Phase-II-Studie getestet. Sicherheitsaspekte dürften bei diesem Therapieansatz eine große Rolle spielen, denn es stellt sich die Frage, wie gut ein Organ, aus dem größere Mengen von Amyloid-Ablagerungen entfernt wurden, danach noch funktioniert.


