 |  | Jennifer Evans |
 |
14.01.2025 09:00 Uhr |

Bei einem Fälschungsverdacht ist es wichtig, die betroffene Schachtel zu separieren und das eigene Vorgehen genau zu dokumentieren. / © Adobe Stock/Dragana Gordic
Neben der Verifikation über das Securpharm-System sind Apothekerinnen und Apotheker angehalten, eine Rx-Arzneimittelpackung vor Abgabe zusätzlich per Sichtkontrolle zu prüfen. Denn das Fälschungsschutz-System besteht neben dem DataMatrix Code, der sich über die Datenbank überprüfen lässt, auch aus einem Erstöffnungsschutz. Auffällig wäre zum Beispiel, wenn dieser beschädigt oder die Packung bereits geöffnet ist. Ebenso skeptisch machen sollte ein ungewöhnlicher Geruch, ein untypisches Gewicht oder, wenn sich etwas lose in der Schachtel bewegt. Durch ihren täglichen Umgang mit den Medikamenten können Apothekerinnen und Apotheker bereits einiges erkennen.
Wachsam sein sollte das Apothekenteam darüber hinaus, wenn die Packungsdaten leicht abgewandelt sind. Basiert etwa ein neuer, aber falscher DataMatrix-Code teilweise auf dem einer echten Packung, kommt es im System zu einem Alarm aufgrund eines technischen Fehlers. Eine solche Schachtel kann im hektischen Alltag schnell einmal durchrutschen, aber durchaus eine Fälschung sein. Des Weiteren ist es möglich, dass eine Packung einen Import vortäuscht, der außerhalb des Schutzsystems eine Ausnahme darstellt, aber faktisch ebenfalls eine Fälschung sein könnte.
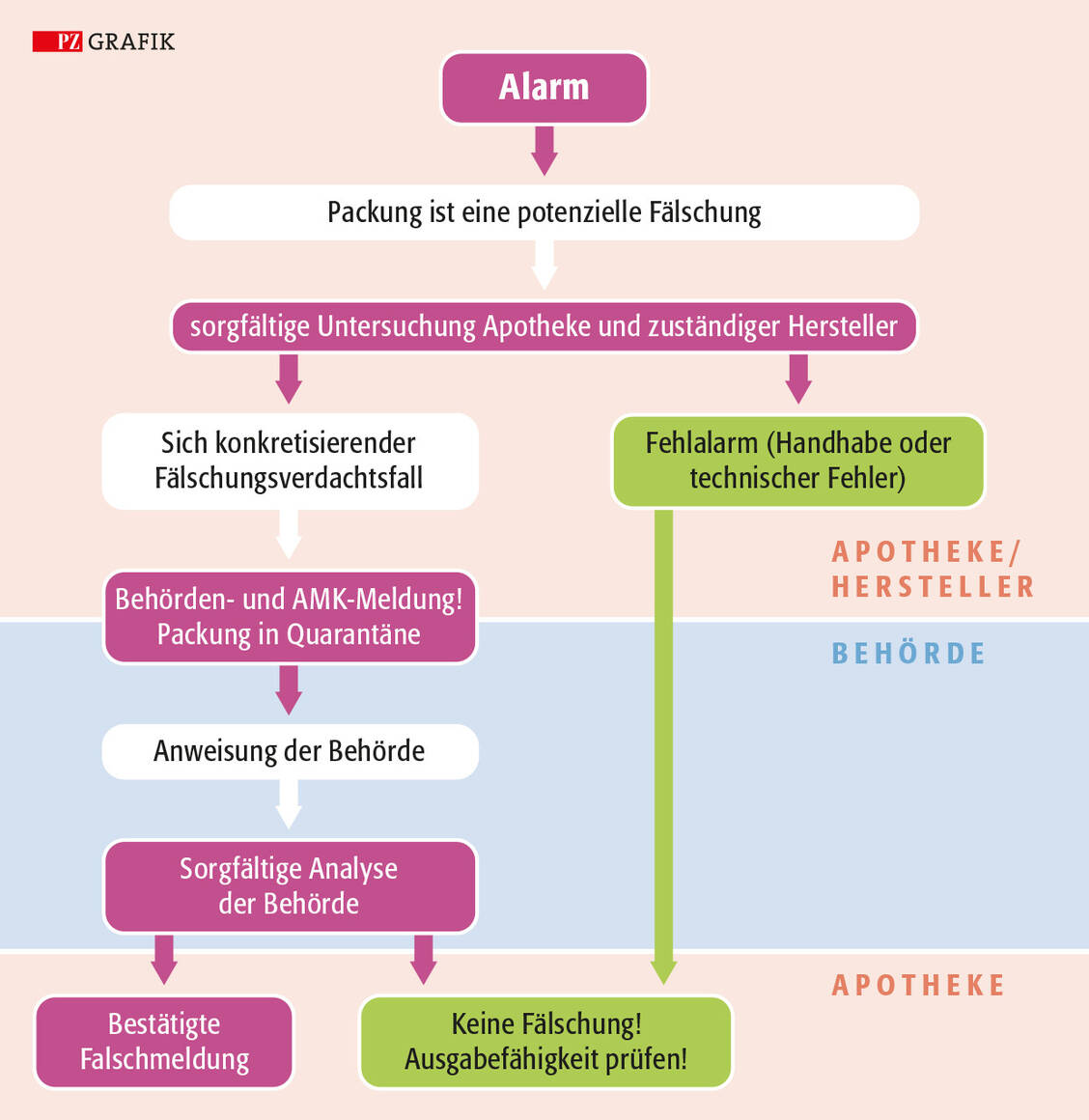
Im Zweifelsfall gilt es, nicht zu zögern. Liegt ein konkreter Verdacht vor, muss die Apotheke diesen ohnehin sofort melden – bei der zuständigen Aufsichtsbehörde sowie bei der Arzneimittelkommission der Deutschen Apotheker (AMK). Bei unklaren Fehlermeldungen erlaubt der Gesetzgeber eine Frist von sieben Tagen, um gegebenenfalls eine Rückmeldung vom Hersteller abwarten zu können. Tendenz bleibt aber: je schneller eine Offizin bei einem Alarm handelt, desto besser.
Grundsätzlich gilt: Für die Überwachung des legalen Vertriebswegs von Arzneimitteln sind die Arzneimittelüberwachungsbehörden der Länder zuständig. Und für die Verfolgung von Straftaten Polizei und Zollfahndungsdienste im Auftrag der Staatsanwaltschaft.
Derzeit wird ein Alarm aber nicht automatisch an die Aufsichtsbehörde weitergeleitet. Das liegt daran, dass der Gesetzgeber unter anderem in der Apothekenbetriebsordnung (ApBetrO) festgelegt hat, dass der Alarm zunächst untersucht werden soll, bevor er offiziell gemeldet wird. Sprich: Im konkreten Fälschungsverdacht reicht es also nicht aus, den Alarmstatus im Alarm-Management-System auf »Eskaliert« zu setzen, um den Meldeverpflichtungen nachzukommen.
Zuständig ist in der Regel die jeweilige regionale Aufsichtsbehörde, bei der eine Apotheke auch ihre Betriebserlaubnis eingeholt hat. Bei einem konkreten Fälschungsverdacht sind zusätzlich mehrere offizielle Stellen involviert. Unterscheiden wird zwischen der erstinvolvierten, der zuständigen Landesbehörde und der zuständigen Bundesoberbehörde. Die beiden ersten können identisch sein und leiten einen Fälschungsverdachtsfall an die Bundesoberhörde weiter. Die Prüfung, ob es sich um eine tatsächliche Fälschung handelt, kann jedoch einige Zeit in Anspruch nehmen. Denn auch die Behörden müssen zunächst die labortechnische Untersuchung abwarten.
Sofern es sich dann um gefälschte, nicht zugelassene oder nicht registrierte Fertigarzneimittel handelt, informieren die Behörden entsprechend Polizei, Zoll und Staatsanwaltschaft und legen einen Datensatz in der Fälschungsdatenbank an. Außerdem wird ein sogenanntes Rapid Alert Notification (RAN) Formular in englischer Sprache erstellt. Handelt es sich um zentrale Zulassungen und Parallelvertrieb benachrichtigen die Bundesoberbehörden auch die Europäische Arzneimittelagentur – EMA. Zudem fällt das Informieren der Fachpresse und der Öffentlichkeit in ihren Zuständigkeitsbereich.
© PZ
Bei Fälschungsverdacht muss die Apotheke eine Packung sofort separieren und entsprechend kennzeichnen etwa mit der Notiz »nicht zum Verkauf bestimmt« oder aber mit einem Fälschungsverdacht-Aufkleber. Es bietet sich an, für diese Präparate in der Offizin einen festen Aufbewahrungsort einzurichten, damit das gesamte Team weiß, dass die Packung nicht abgabefähig ist. Dieser Ort sollte sich dauerhaft an derselben Stelle befinden und sich nicht ständig ändern. Dort muss die Schachtel so lange lagern, bis klar ist, wie das weitere Vorgehen aussieht.
Vor Schutz gegen unbefugten Zugriff ist eine Fälschungsverdacht-Schachtel in einem abschließbaren Schrankfach aufzuheben. Auch dieses muss beschriftet sein, um Verwechslungen mit anderen Arzneimitteln auszuschließen. Ist ein Medikament oder ein Ausgangstoff nicht mehr verkehrsfähig, muss der Apothekenleiter oder ein Teammitglied diesen zurückgeben oder vernichten. In jedem Fall sollte eine Offizin den gesamten Prozess dokumentieren, inklusive der Meldungen an die Behörden.
Kommt es zu einem Abgabeverbot einer Arzneimittelpackung, dann handelt es sich um einen Sachmangel. Gemäß Bürgerlichen Gesetzbuch (BGB) haftet dafür der Verkäufer, also der Großhändler – sofern die Apotheke den Fehler nicht selbst verursacht hat. Allerdings muss die Apotheke den Großhändler unverzüglich über einen Fälschungsverdacht informieren. Andernfalls verliert sie laut Handelsgesetzbuch (HGB) ihre gesetzlichen Gewährleistungsrechte.
Ob es dann bei einer solchen Mängelanzeige aufgrund eines Fälschungsverdacht später eine Rückzahlung oder neue Ware für die Apotheke gibt, hängt vor allem von den individuell vereinbarten Vertragsbedingungen zwischen Offizin und Großhändler ab.
1. Wenn der Hersteller die Daten einer Packung nicht hochgeladen hat, sieht es im Alarm-Management-System bei der Verifzierung oder Abgabe aus wie ein technischer Alarm.
2. Wenn der Hersteller oder Großhändler versehentlich die Packung ausbucht, ist das für die Apotheke nicht ersichtlich, da nur die eigene Transaktionshistorie überprüfbar ist.
3. Wenn eine benachbarte Apotheke den Kollegen ausgeholfen hat, sieht die Apotheke nur, dass die Packung bereits »inaktiv« ist.
4. Wenn der Packungsstatus in der Apotheke nicht setzbar ist, sprich durch nur durch andere Akteure möglich ist, zum Beispiel bei einem Muster.
5. Wenn die Packung nicht für den europäischen Markt serialisiert ist, hilft nur der Abgleich zu den Ausnahmegenehmigungen des BfArMs, der AMK oder des Herstellers sowie eine Prüfung anhand der eigenen Erfahrung beziehungsweise Sinneseindrücke.


