 |  | Gerd Bendas |
 |
14.07.2022 11:00 Uhr |

Physio- und oft auch Ergotherapie gehören zum Therapiespektrum bei Multipler Sklerose immer dazu. Die medikamentösen Optionen haben in den letzten Jahren erhebliche Fortschritte gemacht. / Foto: Adobe Stock/herraez
Die Multiple Sklerose (MS) ist eine autoimmun induzierte neurologische Entzündungserkrankung, von der gegenwärtig in Deutschland etwa 250.000 Menschen betroffen sind. Die Zahl der jährlich neu diagnostizierten Patienten wird mit etwa 10.000 angegeben. Die Erkrankung tritt meist zwischen dem 20. und 40. Lebensjahr auf, wobei zunehmend über ältere Neuerkrankte berichtet wird. Frauen weisen eine höhere Inzidenz auf.
Charakteristisch für eine Autoimmunerkrankung: Die Pathologie liegt in einer Deregulierung des Immunsystems und einem immunologischen Angriff auf körpereigenes Gewebe begründet. In einer meist schubförmigen Verlaufsaktivität invadieren autoreaktive T- und B-Lymphozyten in das zentrale Nervensystem (Gehirn und Knochenmark) und induzieren eine Demyelinisierung der Nervenbahnen (Abbildung 1). Daraus resultieren entzündliche Prozesse und axonale Schäden (1), die sich während eines Krankheitsschubs in vielfältigen neurologischen Ausfallerscheinungen manifestieren.
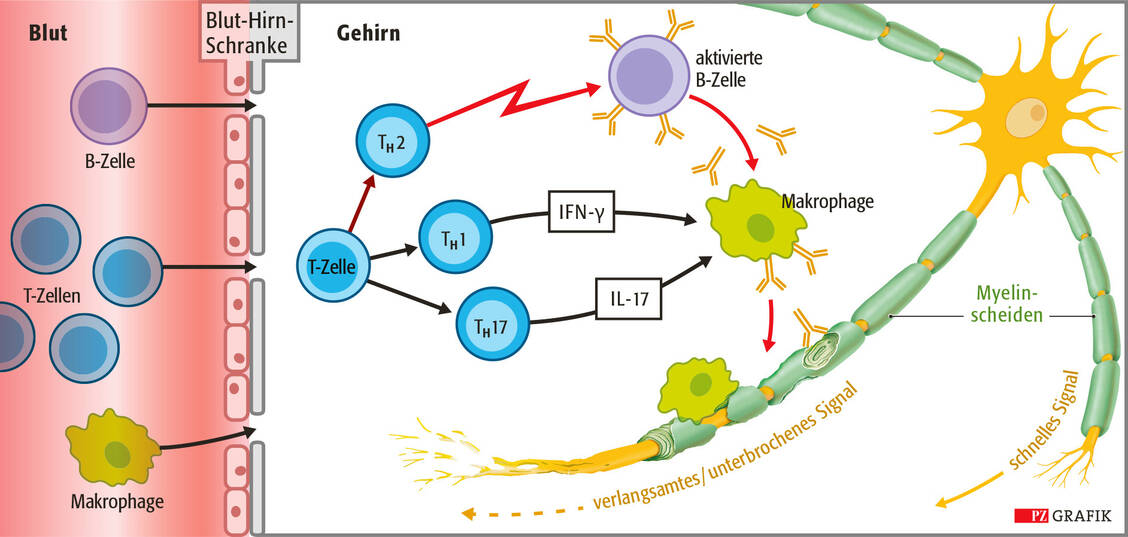
Abbildung 1: Der Pathomechanismus bei MS ist durch die Invadierung von T- und B-Lymphozyten sowie Makrophagen über die Blut-Hirn-Schranke ins Gehirn geprägt. Dort erfolgt eine weitere Aktivierung und Differenzierung der Lymphozyten, in deren Folge aktivierte Makrophagen die Axone der Nervenzellen angreifen. / Foto: PZ/Stephan Spitzer
Die in einem akuten Schub entstehenden »multiplen« Entzündungsherde verheilen teilweise wieder unter Narbenbildung, »sklerosieren« also in einer Remissionsphase. Die Symptomatik der MS ist extrem vielfältig und hängt ab von der Intensität der Schädigung und deren Lokalisation im Gehirn. Die häufigsten Symptome umfassen Seh- und Sprachstörungen, Taubheitsempfindungen, Gangunsicherheiten sowie Lähmungen.
Autoimmune Erkrankungen gelten immer noch als unheilbar. Bei der MS besteht eine gewisse genetische Prädisposition. Im Fokus stehen die HLA-(Humane Lymphozyt Antigen-)DR15-Haplotyp-Modifikationen (2), die eine fehlerhafte Antigenpräsentation induzieren. Diese können zusammen mit externen Faktoren zur Fehlregulation des Immunsystems führen.

Die Symptomatik der MS ist extrem vielfältig. Am häufigsten sind Seh- und Sprachstörungen, Taubheitsempfinden, Gangunsicherheit und Lähmungen. / Foto: Adobe Stock/doucefleur
Seit vielen Jahren ist bekannt, dass virale Infektionen offensichtlich mit der Induktion der MS korrelieren. Daher hat kürzlich eine beweiskräftige Studie zum Zusammenhang der MS-Inzidenz mit einer früheren Infektion mit Epstein-Barr-Viren (EBV) große Aufmerksamkeit auf sich gezogen. Diese epidemiologischen Daten beweisen, dass eine Infektion mit EBV das Risiko, an MS zu erkranken, um das 32-Fache gegenüber EBV-negativen Probanden erhöht (3).
Mit zunehmenden Einblicken in die vielfältigen, auch immunologisch steuernden Funktionen der intestinalen Mikrobiota (Darmbakterienflora) werden auch Zusammenhänge mit der MS-Erkrankung deutlich. Fakt ist, dass die Patienten eine veränderte Zusammensetzung der Mikrobiota gegenüber Gesunden aufweisen, was zum Ungleichgewicht zwischen aktivierten T-Zellen (TH17) und immunsuppressiven Treg-Zellen beitragen kann. Studien belegen, dass durch die mikrobielle Imbalance bestimmte Regulatorstoffe, zum Beispiel Propionsäure, fehlen. Eine Patientenstudie mit therapeutisch immunmodulierten MS-Erkrankten zeigte eindrucksvoll, dass die orale Supplementierung von Natriumpropionat die Immunbalance und die Symptomatik positiv beeinflusst (4). Der genaue Mechanismus ist unbekannt.
Als ein weiterer externer Faktor erhöhter MS-Inzidenz ist Rauchen epidemiologisch klar belegt. Auch ein Mangel an Vitamin D wird seit vielen Jahren diskutiert, womit sich das Nord-Süd-Gefälle in der Erkrankungsstatistik der MS erklären lässt. Eine aktuelle deutsche Studie zeigt an zwei geografisch unterschiedlichen Patientenkohorten die Zusammenhänge von Sonneneinstrahlung und Vitamin-D-Spiegeln mit der Schwere der Krankheitsverläufe, dem Risiko für Rückfälle und der langfristigen Behinderungsprogression (5). Solche Befunde unterstützen eine kontrollierte Vitamin-D-Supplementierung (14.000 I.E. täglich). Die in den letzten Jahren diskutierte Ultrahochdosistherapie (mehr als 100.000 I.E. täglich) ist aufgrund vielfältiger Risiken zunehmend passé.
Es werden drei Verlaufsformen unterschieden, wobei das sogenannte klinisch isolierte Syndrom (CIS) oft ein Anfangsstadium der Krankheit markiert. Bei etwa 80 Prozent der Neuerkrankten tritt eine schubförmig remittierende MS (relapsing remitting; RRMS) auf: Entzündungsschübe mit mindestens 24-stündiger Symptomatik klingen dann wieder (nahezu) vollständig ab. Verschlechtern sich die Ausfallerscheinungen und damit die Beschwerden der Patienten über sechs Monate kontinuierlich, wird dies als sekundär progrediente MS (SPMS) bezeichnet. Viele Patienten erleiden nach längerer Erkrankungszeit einen Übergang von der RRMS zur SPMS.
Bei 10 bis 15 Prozent der Patienten verstärkt sich die Krankheitsintensität kontinuierlich ohne ausgesprochene Schubaktivität; dies wird als primär progrediente Verlaufsform (PPMS) bezeichnet.
Die therapeutisch verwendeten Arzneistoffe wirken mit verschiedensten Ansatzpunkten immunmodulierend oder immunsuppressiv. Im Februar 2021 wurde die von der Deutschen Gesellschaft für Neurologie aktualisierte und erweiterte S2k-Leitlinie zur Therapie und Diagnostik der MS veröffentlicht (6). Ziel dieser konsensbasierten Leitlinie ist eine stärker individualisierte und evidenzbasierte Behandlung.
Neu gegenüber dem bis dato geltenden starren Stufenschema ist eine Festlegung von drei Wirksamkeitskategorien, denen die verlaufsmodifizierenden MS-Wirkstoffe zugeordnet werden. Die Zuordnung basiert auf der in klinischen Studien bewiesenen Reduktion der entzündlichen Schubraten (Tabelle). Diese beträgt bei Wirkstoffen der Kategorie 1 etwa 30 bis 50 Prozent und der Kategorie 2 circa 50 bis 60 Prozent versus Placebo, während bei Wirkstoffen der Kategorie 3 von einer über-60-prozentigen Reduktion versus Placebo oder 40 Prozent gegenüber Kategorie-1-Arzneistoffen ausgegangen wird.
| Kategorie | Relative Reduktion der Schubrate (verglichen mit Placebo) | Wirkstoffe |
|---|---|---|
| 1 | 30 bis 50 Prozent | Beta-Interferon, PEG-InterferonDimethylfumaratGlatirameroideTeriflunomid |
| 2 | 50 bis 60 Prozent | CladribinS1P-Rezeptor-Modulatoren: Fingolimod, Ozanimod, Ponesimod |
| 3 | > 60 Prozent oder> 40 Prozent verglichen mit Kategorie 1 | AlemtuzumabCD20-Antikörper: Ocrelizumab, Rituximab (off Label)Natalizumab |
Diese Einteilung lässt mehr Freiheitsgrade. Obwohl bei Therapiebeginn meist Wirkstoffe der Kategorie 1 genutzt werden, kann der Arzt bei einer erwartet schweren Verlaufsform auch direkt mit höheren Kategorien behandeln. Höhere Kategorie und damit stärkere Wirksamkeit können, müssen aber nicht mit stärkeren Nebenwirkungen einhergehen.
Unverändert bleibt die Therapie akuter Schübe durch die intravenöse Gabe von Methylprednisolon (500 bis 1000 mg täglich) für drei bis fünf Tage. Die Leitlinie eröffnet für jede Kategorie Beispiele möglicher Wechsel- oder Ausstiegsszenarien. Ein in der PZ 49/2021 erschienener Titelbeitrag adressiert dies explizit (7), daher werden diese Aspekte hier nicht näher erläutert. Die Leitlinie klärt zudem, inwieweit eine initial stärkere Therapie langfristig zu einer reduzierten Behinderungsprogression führt (8).
Beta-Interferone
Interferon beta 1a (Avonex®, Rebif®; ein- bis dreimal wöchentlich intramuskulär, i.m., oder subkutan, s.c.) und Interferon beta 1b (Betaferon®; s.c. alle zwei Tage) wirken modulierend auf die Autoimmunreaktion und verhindern so Entzündungsschübe und Progression bei RRMS. Betaferon® und dessen Bioidentical Extavia® können auch bei schubförmiger SPMS eingesetzt werden. Als Wirkmechanismen der Beta-Interferone werden eine Aktivierung antiinflammatorischer Zytokine sowie eine reduzierte T-Zell-Aktivierung durch Studien bestätigt. Plegridy®, ein pegyliertes Interferon beta 1a, ist nur für Erwachsene empfohlen. Es hat eine deutlich verlängerte Halbwertzeit und muss daher nur zweimal monatlich subkutan appliziert werden. Die häufigste Nebenwirkung unter Beta-Interferonen besteht im Auftreten grippeähnlicher Symptome. Die Verordnung (7,8 Millionen DDD 2020) nimmt seit Jahren kontinuierlich ab (9).
Glatirameroide
Das seit 2001 zugelassene Glatirameracetat (Copaxone®, Nachahmerprodukt Clift®) ist ein synthetisches, molekular heterogenes Peptidgemisch (»Glatirameroide«) von etwa 5 bis 9 kDa, das durch einen nicht genau definierten Mechanismus die autoimmunen Entzündungsreaktionen und damit die Schubrate bei RRMS-Patienten senken kann. Copaxone wird subkutan appliziert (20 mg täglich oder 40 mg alle zwei Tage) und ist mit 5,2 Millionen DDD 2020 seit Jahren gleichbleibend akzeptiert (9).
Teriflunomid (Aubagio®)
Teriflunomid ist seit 2013 als oraler Wirkstoff (14 mg täglich) zur Therapie der RRMS zugelassen. Teriflunomid ist bekannt als aktiver Metabolit des Leflunomids aus der Therapie der rheumatoiden Arthritis. Es hemmt die Dihydroorotat-Dehydrogenase in den Mitochondrien und blockiert so die Nukleotid-Synthese. Dies beeinflusst die Proliferationsfähigkeit der Lymphozyten; funktionell zeigt sich dies in einer um etwa 30 Prozent reduzierten Schubrate. Studien belegen die therapeutische Gleichwertigkeit zu den Beta-Interferonen. Mit 4,4 Millionen DDD 2020 weist Teriflunomid deutliche Zuwachsraten auf, wohl auch wegen der vergleichsweise günstigen Kosten (9).
Teriflunomid sollte bei Patienten mit beeinträchtigtem Immunstatus, Leberfunktionsstörungen oder dialysepflichtigen Nierenfunktionsstörungen nicht angewendet werden. In der Schwangerschaft ist es streng kontraindiziert. Zu beachten ist, dass der Wirkstoff einem enterohepatischen Kreislauf unterliegt (Halbwertszeit 19 Tage) und bei einer ungeplanten Schwangerschaft aktiv (Aktivkohle, Cholestyramin) eliminiert werden sollte. Unter der Therapie mit Teriflunomid ist auf eine regelmäßige Kontrolle der Leberwerte und des Blutbilds zu achten. Auf pharmakokinetischer Ebene (Inhibitor von CYP2C8, Substrat von CYP1A2 und 2C19) sind Arzneimittelwechselwirkungen möglich.
Fumarate
Dimethylfumarat (Tecfidera®) ist seit 2014 als orales Therapeutikum (zweimal täglich 240 mg) zur MS-Therapie zugelassen. Der aktive Metabolit Methylfumarat (Abbildung 2) aktiviert unter anderem den Transkriptionsfaktor Nrf2 und wirkt über diesen Signalweg antiinflammatorisch. Mit 7,3 Millionen DDD ist Dimethylfumarat das aktuell am stärksten verschriebene MS-Therapeutikum in Deutschland (9). Eine mit dem Wirkmechanismus einhergehende Lymphopenie kann die Patienten für sekundäre Infektionen sensibilisieren; so wurden bereits Fälle einer progressiv multifokalen Leukenzephalopathie (PML) beschrieben. Daher wurden strenge Auflagen zum Monitoring der Patienten erlassen (Rote-Hand-Brief 11/2020) (10). Als häufige Nebenwirkungen werden das Auftreten eines plötzlichen Hitzegefühls (Flush) und Magen-Darm-Probleme berichtet.
Seit Januar 2022 ist Diroximelfumarat (Vumerity®) in Deutschland zur peroralen Therapie (anfangs zweimal täglich 231 mg, nach sieben Tagen zweimal täglich 462 mg) auf dem Markt (Abbildung 2). Bei gleichem aktiven Metaboliten ist eine analoge Wirksamkeit zu Dimethylfumarat als gegeben anzusehen, jedoch belegen Studien eine deutlich bessere gastrointestinale Verträglichkeit (11). Kritische Stimmen werten diese Zulassung auch als eine Strategie zur Kompensation im Fall von Generika-Zulassungen von Dimethylfumarat.
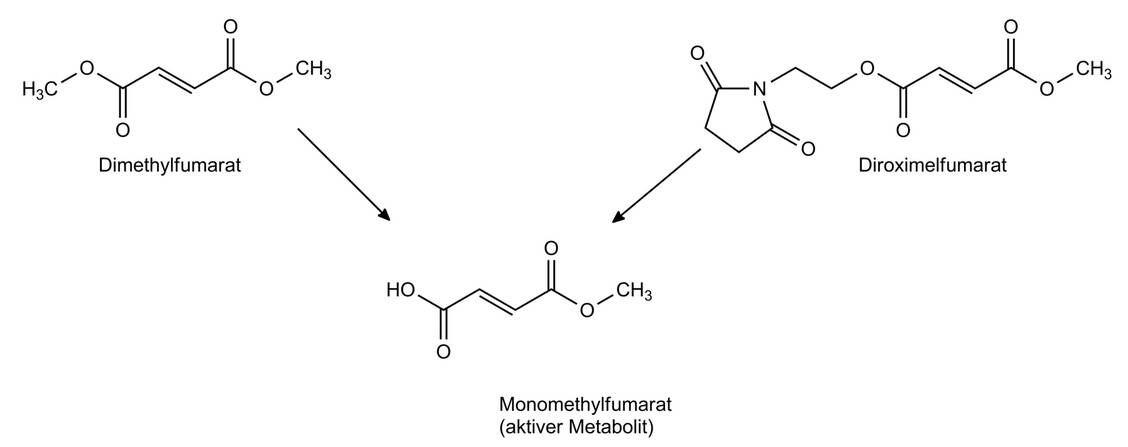
Abbildung 2: Dimethylfumarat und das 2022 neu zugelassene Diroximelfumarat werden durch Esterspaltung in den aktiven Metaboliten Monomethylfumarat umgewandelt. / Foto: Bendas, Wurglics
Cladribin (Mavenclad®) ist ein chloriertes Desoxyadenosin und wirkt als Nukleotid-Antimetabolit. Nach oraler Einnahme wird das Prodrug intrazellulär phosphoryliert und kann dann in die DNA-Synthese eingreifen oder in ruhenden Zellen auf verschiedenen Wegen den Zelltod induzieren. Diese Wirkung ist lang anhaltend und relativ Lymphozyten-spezifisch.

Pluspunkt für die Patienten: Die orale Medikation erleichtert die Adhärenz. / Foto: Adobe Stock/photo 5000
Zu Beginn der Therapie nimmt der Patient für vier bis fünf Tage täglich je nach Körpergewicht 10 oder 20 mg ein. Diese sogenannte orale Kurzzeittherapie wird nach vier Wochen wiederholt, wobei eine kumulative Dosis von 1,75 mg/kg Körpergewicht pro Jahr angestrebt wird. Damit endet die Therapie im ersten Behandlungsjahr. Im zweiten Behandlungsjahr erfolgt eine analoge Einnahme; die zwei folgenden Jahre gelten als behandlungsfrei und sollen für vier Jahre Schubfreiheit garantieren. Etwa 30 Prozent der Patienten sprechen nicht ausreichend an und müssen auf andere Therapeutika wechseln (12).
Die mit der Wirkung einhergehende Lymphopenie verursacht eine Immunsuppression und damit eine höhere Anfälligkeit für Infektionen. Aktive, insbesondere virale Infektionen stellen eine Kontraindikation dar. Der Wirkstoff wird als gut verträglich beschrieben; zwei und sechs Monate nach Therapiebeginn sollte eine Kontrolle der Lymphozytenzahl erfolgen.
Bei der Markteinführung 2011 war Fingolimod (Gilenya®) der erste oral wirksame Arzneistoff in der MS-Therapie. Mittlerweile gibt es drei Nachfolgepräparate, sogenannte »Imode« (Abbildung 3).
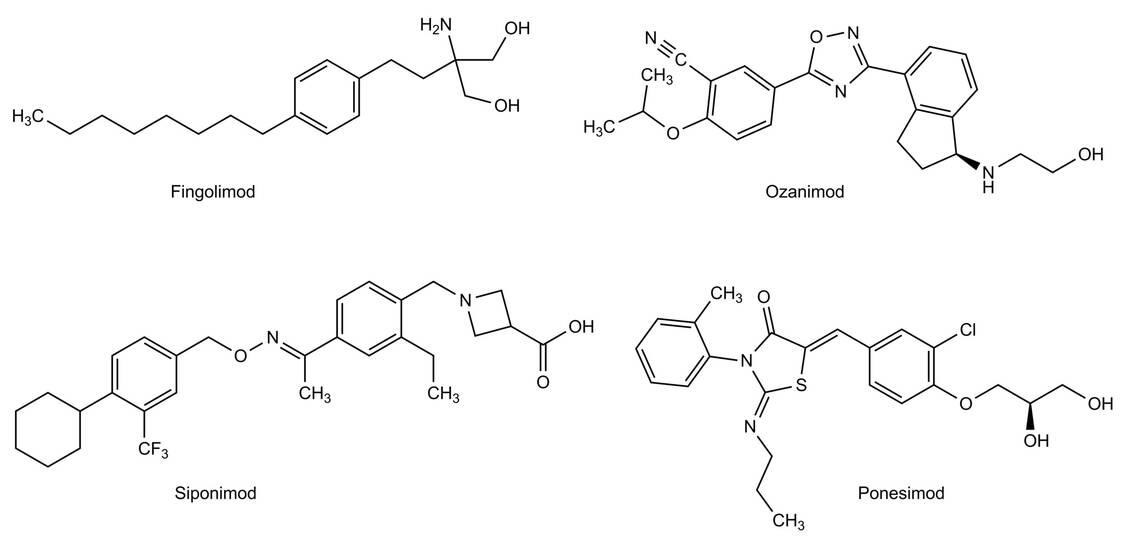
Abbildung 3: Struktureller Vergleich der Sphingosin-1-Phosphat-Rezeptor-(S1PR-)Modulatoren, der sogenannten »Imode« / Foto: Bendas, Wurglics
Fingolimod wird metabolisch phosphoryliert und agiert als Phosphat strukturhomolog zum Lipidmediator Sphingosinphosphat an dessen Rezeptoren. Von den verschiedenen Sphingosin-1-Phosphat-Rezeptoren (S1PR) ist im Kontext der MS-Therapie der Subtyp 1 auf Lymphozyten bedeutsam, denn dieser vermittelt deren Übergang aus den lymphatischen Organen in Entzündungsgebiete im ZNS. Fingolimod fungiert als funktioneller Antagonist an S1PR1, induziert dessen Downregulation und hemmt so den Lymphozytentransfer. Bereits nach der ersten Applikation (0,5 mg täglich) tritt eine drastische Reduktion zirkulierender Lymphozyten auf. Interessanterweise sind die peripheren Lymphozytenfunktionen (Memory- und Effektor-Funktion) deutlich weniger betroffen.
Fingolimod (4,9 Millionen DDD in Deutschland 2020) senkt die Schubrate um mehr als 50 Prozent. Allerdings ist bei dieser immunsuppressiven Therapie einiges zu beachten. Einerseits resultiert eine erhöhte Infektionsanfälligkeit der Patienten, unter anderem für virale Erreger. Aktive Infektionen wie Hepatitis oder Tuberkulose sind klare Kontraindikationen. Fingolimod kann durch Modulation der S1PR des Subtyps 3 auf Zellen des Reizleitungssystems des Herzens kardiale Funktionen, insbesondere zu Therapiebeginn beeinflussen, was eine EKG-Überwachung und Blutdruckkontrolle erfordert. Für Patienten mit kardialen Vorschädigungen ist das Medikament kontraindiziert, ebenso in Schwangerschaft und Stillzeit. Die Leberfunktion solle eng überwacht werden, hieß es in einem Rote-Hand-Brief 2020 (13). Hinsichtlich Arzneimittelwechselwirkungen ist eine Metabolisierung von Fingolimod durch CYP4F2 sowie potenzielle Interaktionen mit CYP3A4-Hemmern zu berücksichtigen.
Siponimod (Mayzent®) ist ein weiterentwickelter S1PR-Modulator, der Anfang 2020 als erste orale Therapieoption für Patienten mit sekundär progredienter Erkrankung (SPMS) eine Zulassung erhielt. Anders als bei Fingolimod wird hier die Modulation von sowohl Subtyp 1 als auch 5 des S1PR beschrieben. Siponimod wird dominant über CYP2C9 metabolisiert, was eine Genotypisierung der Patienten und gegebenenfalls Dosisanpassung erfordert. Hinsichtlich kardialer Nebenwirkungen, Leberfunktionskontrolle und Schwangerschaft gelten die bei Fingolimod getroffenen Erklärungen.
Die monoklonalen Antikörper gehören zu den wirksamsten MS-Therapeutika, müssen aber meist intravenös appliziert werden. / Foto: Getty Images/FG Trade
Ozanimod (Zeposia®) wurde als weiterer S1PR-Modulator im Juli 2020 für Erwachsene mit RRMS zugelassen. Für die orale Therapie (täglich 0,92 mg als Kapsel) wird die Dosis in den ersten acht Tagen wie auch bei Siponimod langsam auftitriert. Pharmakokinetische Wechselwirkungen sind mit CYP2C8-Induktoren sowie MAO-Hemmern zu beachten. Patienten sollten eine starke Exposition mit UV-Strahlung ausschließen und die genannten Vorsichtsmaßnahmen berücksichtigen.
Ponesimod (Ponvory®) kam als neuer Vertreter dieser Verbindungsklasse im Juni 2021 für erwachsene RRMS-Patienten auf den Markt. Auch hier wird langsam aufdosiert, bevor an Tag 14 die Tagesdosis von 20 mg erreicht wird. Bei ansonsten vergleichbarem Wirkungs- und Nebenwirkungsprofil unterscheidet sich Ponesimod leicht durch die etwas kürzere HWZ von etwa 33 Stunden.
Die in diese Kategorie eingeordneten hochwirksamen Arzneistoffe sind therapeutische Antikörper, die entweder durch Bindung an Lymphozyten deren immunologische Depletion induzieren oder die Einwanderung von Lymphozyten aus dem Blutstrom in das ZNS unterdrücken (Abbildung 4).
Natalizumab (Tysabri®) ist ein Antikörper zur Blockade des Adhäsionsmoleküls Integrin α4 auf Lymphozyten, wodurch deren Einwanderung aus dem Gefäßsystem ins ZNS über die Blut-Hirn-Schranke verhindert wird. Es ist seit 15 Jahren in der MS-Therapie gut etabliert (2,2 Millionen DDD 2020, zunehmend) (9) und verringert bei monatlicher intravenöser Applikation von 300 mg die Schubhäufigkeit von RRMS-Patienten stark.
Abbildung 4: Übersicht der in der MS-Therapie angewendeten Immunmodulatoren / Foto: PZ/Stephan Spitzer
Die mit Natalizumab verbundene Immunsuppression kann zum Auftreten einer potenziell tödlichen PML (progressive multifokale Leukenzephalopathie) führen. Daher ist ein enges Monitoring des JC-Virus-Antikörperstatus der Patienten vor und während der Therapie vorgeschrieben.
Alemtuzumab (Lemtrada®), ein Antikörper gegen das auf sowohl B- als auch T-Lymphozyten exprimierte Epitop CD52, folgt dagegen dem erstgenannten Wirkprinzip. Durch seine Bindung an Lymphozyten werden diese für eine Eliminierung durch verschiedene Abwehrmechanismen des Immunsystems markiert. Die damit einhergehende Immunsuppression hemmt die Schubanfälligkeit und die Behinderungsprogression bei der RRMS signifikant.
Der Antikörper wird als Stoßtherapie angewendet, wobei die Patienten an fünf aufeinanderfolgenden Tagen je 12 mg i.v. erhalten. Erst nach zwölf Monaten wird die Therapie mit einer dreimaligen Applikation fortgesetzt. Trotz einer hohen Wirksamkeit wird Alemtuzumab in Deutschland nur eingeschränkt für schwere Verlaufsformen der RRMS verwendet. Eine generell erhöhte Infektionsanfälligkeit der Patienten kann in schweren Fällen auch zu Autoimmunstörungen, zum Beispiel Schilddrüsenerkrankungen oder Nephropathien, führen.
Ocrelizumab (Ocrevus®) ist ein gegen das auf B-Lymphozyten exprimierte Epitop CD20 gerichteter humanisierter Antikörper, der mit analogem Wirkprinzip die immunologische Eliminierung von B-Lymphozyten induziert. Mit seiner Zulassung im Februar 2018 gab es erstmals eine Behandlungsmöglichkeit für die primär progrediente Erkrankungsform (PPMS). In den Zulassungsstudien reduzierte der Antikörper die Behinderungsprogression signifikant. Der Wirkstoff kann auch für schwere Verläufe der RRMS verordnet werden.
Therapeutisch hat Ocrelizumab einen hohen Stellenwert (2,8 Millionen DDD 2020 in Deutschland) erlangt (9). Intravenös werden zweimal je 300 mg im Abstand von zwei Wochen injiziert, gefolgt von 600 mg Einmalgaben alle sechs Monate.
Eine durch CD20-Targetierung induzierte B-Zell-Eliminierung ist nicht neu und wird seit Jahren durch Rituximab bei verschiedenen lymphatischen Erkrankungen und bei rheumatoider Arthritis praktiziert. Rituximab wird seit Jahren off Label bei MS genutzt (14). CD20 erweist sich aus vielerlei Gründen als vorteilhaft. Da dieses Oberflächenepitop nicht in allen Lebensphasen der B-Lymphozyten exprimiert wird (nicht von Stammzellen), bleiben die Fähigkeit zur B-Zell-Rekonstitution und das immunologische Langzeitgedächtnis trotz Therapie erhalten. Für ältere Patienten wird dies aber mit größerer Vorsicht betrachtet und sollte für Über-50-Jährige im Einzelfall erörtert werden.
Ein weiterer Transfer eines CD20-Antikörpers aus der Leukämietherapie ist mit Ofatumumab zu verzeichnen, das als Kesimpta® im September 2021 für Erwachsene mit RRMS zugelassen wurde. Ofatumumab erkennt eine andere Sequenz des CD20-Moleküls, was mit einer erhöhten Markierungseffizienz der B-Zellen und damit Wirkung einhergehen soll (Abbildung 4). Wichtig für die Patienten: Dies ist der erste Antikörper in der MS-Therapie, den sie selbst applizieren können. Dabei wird eine Dosis von 20 mg mittels Fertigpen subkutan in Bauch, Oberschenkel oder Arme in den Wochen 0, 1 und 2 (Initialphase) sowie ab Woche 4 monatlich injiziert (Erhaltungstherapie).
Verschiedene Arzneistoffe befinden sich in fortgeschrittenen Phasen der klinischen Entwicklung für die MS-Therapie. Auf einige Kandidaten soll hier kurz eingegangen werden.
Sehr aussichtsreich und fortgeschritten sind Hemmstoffe der Bruton-Tyrosinkinase (BTK). Die BTK ist ein in B-Zellen zytoplasmatisch lokalisiertes Enzym, durch dessen Aktivierung die B-Zell-Reifung, -Proliferation und Chemokinfreisetzung getriggert werden. Hemmstoffe der BTK wie Ibrutinib werden in der Onkologie zur Behandlung von B-Zell-Lymphomen eingesetzt. Durch die Schlüsselstellung von B-Zellen in der Pathogenese der MS versprechen BTK-Inhibitoren eine kausale Einflussnahme auf den Entzündungsfortgang. Vier Wirkstoffe sind in ihrer klinischen Entwicklung weit fortgeschritten.
Evobrutinib (Merck) befindet sich aktuell in einer Patienten-rekrutierenden Phase-III-Studie zur Behandlung Erwachsener mit RRMS. Tolebrutinib (SAR442168; Sanofi-Genzyme) wird aktuell in zwei Phase-III-Studien bei RRMS und SPMS getestet. Orelabrutinib (Biogen) ist aktuell in Phase II. Während diese drei Inhibitoren eine irreversible Inhibition der BTK durch Bindung an einen Cysteinrest (C483) induzieren, repräsentiert Fenebrutinib (Roche) einen anderen reversiblen Bindungsmodus. Die hohen Erwartungen an diese Verbindungsklasse scheinen durch die bisherigen klinischen Befunde gerechtfertigt. Für genauere strukturelle und klinische Parameter der BTK-Inhibitoren sei auf spezifische Übersichtsartikel verwiesen (15).
Masitinib ist ein Tyrosinkinase-Inhibitor, der aus dem onkologischen (veterinärmedizinischen) Kontext bekannt ist. Der Wirkstoff befindet sich in Phase III der klinischen Entwicklung und soll seine hemmende Wirkung insbesondere auf das angeborene Immunsystem entfalten.
Seit einigen Jahren wird hoch dosiertes Simvastatin als potenzielle Therapieoption untersucht. Nach ersten Befunden, dass dadurch die Atrophie im ZNS reduziert werden kann (16), wird es aktuell in Phase III für die Behandlung von RRMS und SPMS entwickelt (17).
Mit Ublituximab befindet sich ein weiterer CD20-Antikörper in Phase III der klinischen Entwicklung zur Therapie der RRMS.
Neben der verlaufsmodifizierenden Therapie der MS durch Immunmodulation werden auch gezielt die Symptome der Erkrankung adressiert. Der Schwerpunkt liegt dabei auf der Linderung der Spastiken, die in fortgeschrittenen Phasen bei vielen Patienten auftreten. Wirkstoffe mit spasmolytischer und muskelrelaxierender Wirksamkeit werden ergänzend zu Physiotherapien eingesetzt.
Fampridin (Fampyra®) ist als 4-Aminopyridin ein Kaliumkanalblocker, der die Impulsübertragung in geschädigten Nervenarealen verbessern und damit die Motorik positiv beeinflussen soll. Es ist exklusiv für die MS-Therapie zur Verbesserung der Gehfähigkeit zugelassen und wird mit 5,0 Millionen DDD 2020 auch stark verschrieben. Zweimal täglich wird eine Retardtablette (10 mg) geschluckt. Wirksamkeit und Ansprechquote von Fampridin werden jedoch zunehmend kritisch betrachtet (18).
Sehr prominent ist Nabiximols (Sativex®), das als Extrakt aus Cannabis sativa ein standardisiertes Gemisch aus Delta-9-Tetrahydrocannabinol und Cannabidiol darstellt und als Spray oral angewendet wird. Pro Sprühstoß werden 2,7 mg THC und 2,5 mg CBD appliziert. Das Spray unterliegt der Betäubungsmittel-Verschreibungsverordnung. Die Ansprechquote ist moderat (etwa 40 Prozent). Eine Behandlung sollte nach vier Wochen beendet werden, wenn sich die Spastik nicht deutlich bessert. Das Medikament sollte nicht bei Patienten mit Disposition für Schizophrenie, Psychosen oder Persönlichkeitsstörungen angewendet werden.
Baclofen, ein zentralwirksames GABA-Derivat, vermindert den Tonus der Skelettmuskulatur durch Verbesserung der neuronalen Übertragung und wird daher auch zur symptomatischen Therapie der MS eingesetzt.
Auch Botulinumtoxin A (BTX) (Botox®) spielt bei vielfältigen neurologischen Störungen mit spastischer Muskelkontraktion eine große Rolle. Parenteral verabreicht hemmt BTX die periphere Acetylcholin-Freisetzung an präsynaptischen Nervenenden und hemmt so lang anhaltend die neuromuskuläre Übertragung. Speziell bei MS-Patienten soll Botox eine Harninkontinenz mit neurogener Detrusorhyperaktivität dämpfen.
Tizanidin (Sirdalud®) ist ein α2-Adrenozeptor-Agonist und wird als zentral wirksames Muskelrelaxans mit individueller Dosierung (2 bis 24 mg/d) eingesetzt. Es gilt als Alternative zu Baclofen (19).

Foto: ABDA
Grundsätzlich zählen MS-Patienten nicht zu einer Risikogruppe, die bei einer SARS-CoV-2-Infektion vermehrt unter schweren Verlaufsformen der Erkrankungen leidet. Es zeigte sich auch, dass eventuelle Defekte in der Blut-Hirn-Schranke bei MS-Patienten keine zentrale virale Invasion oder andere Krankheitsverläufe begünstigen. Bei der Therapie einer Covid-19-Erkrankung muss patientenindividualisiert entschieden werden.
Klar ist, dass unter Beta-Interferonen, Glatirameroiden, Dimethylfumarat, Teriflunomid sowie Natalizumab nicht von einem erhöhten Risiko schwerwiegender Lungenerkrankungen auszugehen ist. Bei Therapie mit den S1PR-Modulatoren besteht ein leicht erhöhtes Risiko, wobei dies im Regelfall nicht zum Therapieabbruch führen, sondern eher die Expositionsprophylaxe intensivieren sollte. Die CD20-Antikörper, Alemtuzumab sowie Cladribin erhöhen das Risiko insbesondere bei Therapiebeginn, sodass hier eine zeitliche Verschiebung der Therapie oder ein Ausweichen auf andere Wirkstoffe empfohlen wird.
Durch die lebenslang notwendige Behandlung der MS stellen sich oftmals Fragen, inwieweit dies mit verschiedenen Lebensumständen zu vereinbaren ist. Beispielhaft soll hier auf einige Aspekte eingegangen werden.
Eine MS-Therapie ist auch in einer Schwangerschaft möglich, wobei die Frau bereits vorher therapeutisch gut eingestellt sein sollte. Jede Therapiefortführung muss individualisiert betrachtet werden. Einige Arzneistoffe sind aufgrund ihres Wirkmechanismus klar kontraindiziert: Teriflunomid, die S1PR-Modulatoren, Cladribin, Fumarate sowie die CD20-Antikörper.
Neuere Studien belegen, dass Beta-Interferone in Abstimmung mit dem behandelnden Neurologen angewendet werden können; 2019 erfolgte diesbezüglich eine Zulassungserweiterung durch die EMA. Dies deckt sich mit den Empfehlungen der Institution Embryotox (20). Auch Natalizumab gilt bei enger Kontrolle als anwendbar. Die Fortführung einer Therapie mit Alemtuzumab scheint akzeptabel, wenn dies zwingend erforderlich ist. Gleiches gilt für die Glatirameroide.
MS-Erkrankungen gelten nicht als Kontraindikation für Impfungen. Totimpfstoffe können jederzeit appliziert werden. Bei Lebendimpfstoffen sollte auf den Grad der Immunsuppression der Patienten geachtet und individuell entschieden werden. Dass durch Immunreaktionen infolge von Impfungen MS-Schübe ausgelöst werden, ist nicht belegt (21).
Gerd Bendas studierte Pharmazie an der Universität Halle, schloss mit dem Diplom ab und wurde 1994 promoviert. Im Jahr 2000 erfolgte die Habilitation für das Fachgebiet Pharmazeutische Chemie. Seit 2003 hat er die Professur für Pharmazeutische Chemie an der Universität Bonn inne. Seine Forschungstätigkeit liegt schwerpunktmäßig auf der Untersuchung der molekularen Mechanismen der Metastasierung und der Chemoresistenz von Tumoren sowie therapeutischen Strategien zu deren Inhibition. Aktuell wird die Möglichkeit zur Hemmung von Chemokinen als Ansatzpunkt einer Einflussnahme auf Entzündungs- sowie Metastasierungsprozesse untersucht.



