 |  | Robert Fürst und Ilse Zündorf |
 |
06.08.2020 11:00 Uhr |
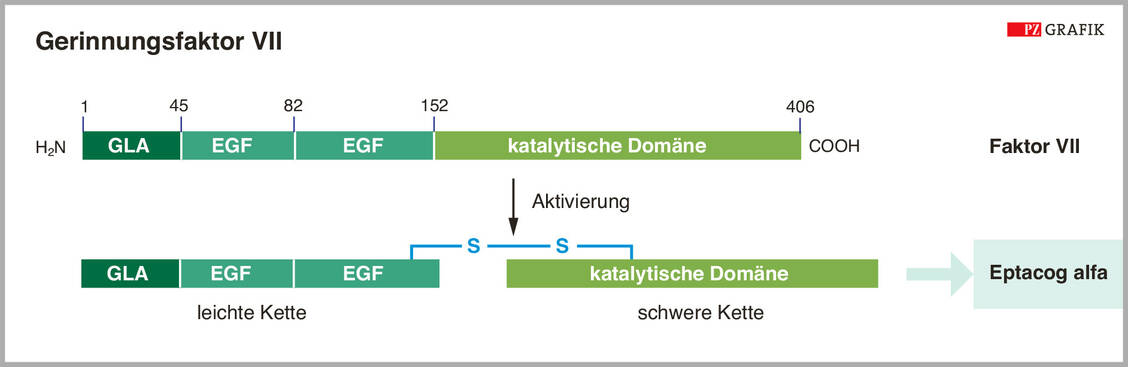
Abbildung 4: Schematische Darstellung des Gerinnungsfaktors VII (oben). Der Arzneistoff Eptacog alfa ist ein bereits aktivierter FVII. / Foto: Grafik: PZ/Spitzer, modifiziert nach I. Zündorf; Dingermann, T., et al., 2019
Ähnlich wie andere Gerinnungsfaktoren wird der Faktor VII in der Leber gebildet und Vitamin-K-abhängig an verschiedenen Glutaminsäure-Resten γ-carboxyliert und zudem noch glykosyliert. Sezerniert wird FVII als einkettiges, 406 Aminosäuren langes Protein (Abbildung 4).
Für die Aktivierung muss das Zymogen von FXa, Thrombin, FIXa oder FXIIa geschnitten werden. Anschließend liegt FVIIa in Form einer leichten und einer schweren Proteinkette vor, die über eine Disulfidbrücke verbunden sind. Nur etwa 1 Prozent des Faktors ist unter physiologischen Bedingungen aktiviert und hat dann eine Halbwertszeit von nur zwei Stunden.
Um bei Operationen nicht übermäßig viel Blut zu verlieren, müssen Patienten mit Hämophilien die fehlenden Gerinnungsfaktoren substituieren. / Foto: Imago/Blickwinkel
Bei Eptacog alfa handelt es sich um den bereits aktivierten FVII (Tabelle 3). Für dieses Präparat wird die cDNA des Gerinnungsfaktors in BHK-Zellen exprimiert. Die Aktivierung des Zymogens findet bereits während der Aufreinigung des Proteins über Ionen-Austausch-Chromatografie statt. Zugelassen ist Eptacog alfa zur Behandlung und Prophylaxe von Blutungen im Zusammenhang mit chirurgischen oder invasiven Eingriffen bei Patienten mit angeborener Hämophilie, die bereits Hemmkörper aufweisen oder bei denen die Gefahr einer Hemmkörperbildung besteht.
Für rekombinanten FVII wurden mittlerweile ebenfalls die gängigen Maßnahmen zur Verlängerung der Halbwertszeit getestet. Diese befinden sich zum Teil in unterschiedlichen Stadien der klinischen Entwicklung.
Die Geschichte der Hämophilie-Therapie zeigt sehr eindrucksvoll die Entwicklung der Biotechnologie und Molekularbiologie. Standen anfangs nur sehr grobe Methoden zur Verfügung, werden mittlerweile ausgeklügelte Moleküle designt, die die Therapie angenehmer und sicherer machen.
Bald könnten auch Gentherapievektoren zum Einsatz kommen, die die ständigen Injektionen der Gerinnungsfaktoren in klinischen Studien bereits für einige wenige Jahre überflüssig gemacht haben. Jedoch sind die normalen Substitutionstherapien bereits sehr kostspielig und die Gentherapien werden noch um einiges teurer werden. Dabei sollte nicht vergessen werden, dass etwa 70 Prozent der Hämophilie-Patienten weltweit wegen der immensen Kosten keinen Zugang zu einer Therapie haben.
Professor Dr. Robert Fürst studierte Pharmazie und erhielt 2001 die Approbation als Apotheker. Anschließend folgten Promotion und Habilitation (2011) im Fach Pharmazeutische Biologie an der Ludwig-Maximilians-Universität München. Seit Ende 2012 hat Professor Fürst die W3-Professur für Pharmazeutische Biologie im Institut für Pharmazeutische Biologie der Goethe-Universität Frankfurt am Main inne. Seit 2016 ist er Geschäftsführender Direktor des Instituts für Pharmazeutische Biologie, seit 2017 Prodekan des Fachbereichs Biochemie, Chemie und Pharmazie. Sein Forschungsschwerpunkt sind die molekularen Wirkmechanismen von Naturstoffen.
Dr. Ilse Zündorf studierte Biologie von 1984 bis 1990 an der Universität Erlangen. Nach einem Forschungsaufenthalt an der Universität of Kentucky, Lexington, USA, wurde sie 1995 am Institut für Pharmazeutische Biologie der Universität Frankfurt promoviert. Zunächst als Akademische Rätin, seit 2001 als Akademische Oberrätin arbeitet sie am Institut für Pharmazeutische Biologie der Goethe-Universität Frankfurt. Ihre Forschungsthemen betreffen Herstellung und Charakterisierung monoklonaler Antikörper, Herstellung und Modifikation rekombinanter Antikörperfragmente sowie die Etablierung von zellulären Testsystemen zur Wirkstoffsuche.
Dingermann, T., Winckler, T., Zündorf, I., Gentechnik – Biotechnik. 3. Aufl., Wiss. Verlagsges. Stuttgart 2019.
Ingram, G. I., The history of haemophilia. J. Clin. Pathol. 29 (1976) 469-479.
Mannucci, P. M., Hemophilia therapy: the future has begun. Haematologica 105 (2020) 545-553.
Peters, R., Harris, T., Advances and innovations in haemophilia treatment. Nat. Rev. Drug Discov. 17 (2018) 493-508.
Key, N. S., Inhibitors in congenital coagulation disorders. Br. J. Haematol. 127 (2004) 379-391.
Giangrande, P., Haemophilia B: Christmas disease. Expert Opin Pharmacother. 6 (2005) 1517-1524.
Liu, J., et al., Improved expression of recombinant human factor IX by co-expression of GGCX, VKOR and furin. Protein J. 33 (2014) 174-183.
Blair, H. A., Emicizumab: A Review in Haemophilia A. Drugs. 79 (2019) 1697-1707.
Lenting, P. J., et al., Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood 130 (2017) 2463-2468.
Spadarella, G., et al., Paradigm shift for the treatment of hereditary haemophilia: Towards precision medicine. Blood Rev. 39 (2020) 100618.
Weyand, A. C., Pipe, S. W., New therapies for hemophilia. Blood 133 (2019) 389-398.
Balkaransingh, P., Young, G., Novel therapies and current clinical progress in hemophilia A. Ther Adv Hematol. 9 (2018) 49-61.
Pedersen, A. H., et al., Autoactivation of human recombinant coagulation factor VII. Biochemistry 28 (1989) 9331-9336.
Jurlander, B., et al., Recombinant activated factor VII (rFVIIa): characterization, manufacturing, and clinical development. Semin Thromb Hemost. 27 (2001) 373-384.
Salas, J., et al., Enhanced Pharmacokinetics of Factor VIIa as a Monomeric Fc Fusion. Thromb Res. 135 (2015) 970-976.
Sandberg, H., et al., Structural and functional characteristics of the B-domain-deleted recombinant factor VIII protein, r-VIII SQ. Thromb Haemost. 85 (2001) 93-100.
Fachinformationen zu den verschiedenen, hier genannten Präparaten.


