 |  | Ulrike Holzgrabe |
| | Martina Kinzig |
| | Fritz Sörgel |
 |
04.05.2025 08:00 Uhr |

Die Coronapandemie hat das Leben sehr vieler Menschen radikal verändert. / © Shutterstock/Ocskay Mark
Lebensmittel und Arzneimittel sind nur begrenzt haltbar und das wird in beiden Fällen in weitreichenden Regularien festgelegt. Bei Lebensmitteln gibt es ein Mindesthaltbarkeitsdatum, zum Beispiel bei Joghurt, der danach noch verzehrt werden kann. Anders ist es bei Hackfleisch oder frischem Fisch; diese sind bis zu einem bestimmten Termin zu verbrauchen. Bei Arzneimitteln ist die Sache einheitlich: Der Verbraucher muss sich an das auf der Verpackung aufgedruckte Verfallsdatum halten.
Das wird in den Medien zu Recht regelmäßig in diese Richtung thematisiert. Aber wie kommt es eigentlich zu einem »Haltbarkeitsdatum« für ein Arzneimittel und zu der berechtigten Forderung an den Patienten, dieses unbedingt einzuhalten?
In der Pandemie zeigte sich mit dem Corona-Wirkstoff Nirmatrelvir als wirksamer Bestandteil in dem Präparat Paxlovid® das Problem einer notwendigen Verlängerung der Haltbarkeitsfrist.
Ein ähnliches Problem war im Jahr 2009 mit der Schweinegrippe aufgetaucht, als die große Bevorratung mit Tamiflu® zu riesigen Verlusten geführt hätte, wenn man die Vorräte zeit- und fristgerecht vernichtet hätte. In den Jahren 2011 und 2012 unterstützte der Hersteller Roche die Länder und belegte durch eigene Stabilitätsuntersuchungen, dass Tamiflu statt fünf Jahre lang, wie üblich bei Medikamenten, bis zu sieben bis zehn Jahre, je nach Charge, verwendet werden durfte.
Mit den hier vorgelegten Stabilitätsuntersuchungen zu Nirmatrelvir soll an einem weiteren Beispiel gezeigt werden, wie das Thema einer verlängerten Haltbarkeit in Sonderfällen – und nur solchen – angegangen werden kann. Es sei darauf hingewiesen, dass die Begründung und auch das Ausmaß der Verlängerung der Haltbarkeit von Tamiflu und Paxlovid unterschiedlich waren.

Paxlovid® war das erste oral einzunehmende Medikament für Covid-19-Patienten zu Hause. Damit mussten weniger Patienten ins Krankenhaus. / © IMAGO/Daniel Scharinger
Studien zur Stabilität von Arzneimitteln sind in der Literatur zahlreich. Für viele, aber nicht alle Arzneimittel zeigt sich, dass nach Packungsangabe verfallene Arzneimittel noch den deklarierten Gehalt an Arzneistoff aufweisen (siehe Übersichtsartikel [1, 2, 3, 4] und repräsentative Studien [5, 6, 7, 8]). Der Verbraucher ist so instruiert, dass er in der Apotheke auf neu hergestellte Arzneimittel drängt und die alten nicht mehr verwenden will.
Es gibt unseres Wissens von den Herstellern selbst keine öffentlich zugänglichen systematischen Studien zu chemisch stabilen Arzneistoffen und -mitteln, die über ein Haltbarkeitsdatum von meist fünf Jahren hinausgehen. Eine Untersuchung einer längeren Haltbarkeit würde allerdings keinen unvertretbar großen Aufwand für die Hersteller bedeuten. Sind Arzneimittel nachweislich über die besagten fünf Jahre hinaus lagerfähig und stabil, dann sollte – bei einem besonderen Anlass wie bei Tamiflu – die längere Haltbarkeitsdauer auf neue Medikamentenpackungen vor dem Inverkehrbringen aufgedruckt werden. Auf keinen Fall sollten in der Übergangszeit im Umlauf befindliche Packungen umetikettiert oder gar zurückgerufen werden.
Die Haltbarkeit eines Arzneistoffs als Bulkware oder in der Arzneiform wird durch die Richtlinien des »International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use« Q1A (2R) von 2003 genau geregelt (9). Das heißt: Stabilitätsstudien finden unter definierten und kontrollierten Bedingungen in Bezug auf Temperatur und Luftfeuchtigkeit statt, bevor das Arzneimittel zugelassen werden kann (Tabelle 1). Die so erhobenen Daten sind sehr artifiziell, aber reproduzierbar, sodass die Haltbarkeit respektive das Verfallsdatum genau angegeben und auf der Verpackung vermerkt werden.
Vorausgegangen sind sichtende Untersuchungen unter harscheren, das heißt beschleunigten (Stress-)Bedingungen (thermischer und chemischer Stress sowie Photostabilität; Details siehe [10]) über sehr kurze Zeiträume. Nachfolgend wird die Stabilität unter sogenannten intermediären Studienbedingungen über einen verkürzten Zeitraum geprüft, insbesondere wenn bei beschleunigten Studien eine Veränderung der Qualität beobachtet wurde. Schließlich werden Langzeitstudien für einen Arzneistoff beziehungsweise ein Arzneimittel durchgeführt (Tabelle 1).
| Bedingungen | beschleunigt | intermediär | Langzeit | Routine |
|---|---|---|---|---|
| Temperatur (°C) | 40 ± 2 | 30 ± 2 | 25 ± 2 | 25 ± 2 |
| Feuchte (Prozent) | 75 ± 5 | 65 ± 5 | 60 ± 5 | 60 ± 5 |
| Dauer (Monate) | 6 | 12 | vorgesehene Laufzeit | vorgesehene Laufzeit |
| Prüfintervalle (Monate) | 0, 1, 3, 6 | 0, 1, 3, 6, 9, 12 | 0, 12, 24, 36, 48 (60) | 0, 12, 24, 36, 48 (60) |
Die in Tabelle 1 angegebenen Einlagerungsbedingungen beziehen sich auf eine geplante Zulassung in subtropischem und Mittelmeerklima, also auf die EU, Japan und die USA (Klimazonen I und II). Bei heißerem und trockenem Klima wie in Wüsten (Klimazone III) oder heißerem und feuchtem, also tropischem Klima (Klimazone IV) wie in Afrika, Asien, Südamerika und Ozeanien wird bei 30 °C und 35 oder 65 Prozent relative Feuchte eingelagert.

Die Bedingungen für die Arzneimittellagerung in Apotheken sind strikt vorgegeben; dies dient der Qualitätssicherung. / © Shutterstock/smoxx
Wie lange sind die Prüfzeiten für eine Lagerung bei Raumtemperatur nach GMP-Regeln (Good Manufacturing Practice)? Man rechnet normalerweise mit einer Studiendauer von bis zu fünf Jahren mit Prüfzeitpunkten nach 0, 3, 6, 9, 12, 18, 24, 36, 48 und 60 Monaten – je nach Medikament oder Studienart (11). Das ist ein großer Aufwand, da die Lagerung unter solch definierten Bedingungen in überwachten Klimaschränken stattfinden und nicht nur ein Batch für jeden Prüfzeitpunkt eingelagert werden muss. Bedacht werden muss auch die geplante Lagerungstemperatur.
Die für die Zulassung notwendigen Stabilitätsprüfungen laufen meist während der klinischen Studien, für die der Arzneistoff ja schon zu einem Arzneimittel formuliert sein muss. Sie dauern bis zu fünf Jahre, was zeitlich gut zur Stabilitätsstudiendauer passt. Am Ende der Studien ist klar, ob und wie lange ein Arzneistoff/-mittel stabil ist und daraus ergibt sich das Verfallsdatum. Viele kleine Moleküle können sehr stabil, das heißt länger als fünf Jahre haltbar sein, während zum Beispiel Biosimilars oder auch β-Lactam-Antibiotika eine deutlich kürzere Haltbarkeit haben. Eine generelle Regel für einzelne Arzneistoffklassen kann aber nicht aufgestellt werden.
Nach der Marktzulassung erfolgen routinemäßig jährliche Stabilitätsuntersuchungen über den gesamten Lebenszyklus eines Arzneimittels, also einmal jährlich über die gegebene Haltbarkeitsdauer. Der Hersteller übernimmt keine Verantwortung für die Anwendung eines länger gelagerten Arzneimittels, unter anderem, weil er dafür keine validen Daten zur Stabilität erhoben hat.
Um es vorwegzusagen: Dem Apotheker kommt nicht die Rolle zu, dem Patienten eine mündliche Erlaubnis oder gar Empfehlung zu geben, ein Medikament auch nach dem auf der Verpackung aufgedruckten Verfallstermin zu benutzen. Auch wenn er – durch seine Kenntnisse in der pharmazeutischen Chemie – eine längere als die erlaubte Verwendung gut begründen könnte.
Wie die Beispiele Tamiflu und Paxlovid zeigen, kann in besonderen Situationen die Haltbarkeit aber »von Amts wegen« verlängert werden.
Wie ist die Verlängerung der Haltbarkeit angesichts der regulatorischen Vorgaben möglich? Nicht einfach, obgleich Untersuchungen von Arzneimitteln außerhalb von regelhaften, wie oben beschriebenen Studien durchaus zeigen, dass Arzneimittel länger haltbar sein können. Das Problem bei weit über die Haltbarkeitsdauer hinausgehenden Arzneimitteln ist, dass sie nicht so kontrolliert gelagert wurden, wie es die ICH-Richtlinie Q1A vorsieht, das heißt, es fehlen überall die Aufbewahrungsarten. Dafür sind es Real-Life-Bedingungen und deshalb wertvoll. Das amerikanische und andere internationale Militärs führen entsprechende Studien durch, da man für den Verteidigungsfall eine Menge Arzneimittel vorrätig halten will, die unter normalen Bedingungen nicht oder kaum benötigt werden.
Das amerikanische Verteidigungsministerium hat bereits 1986 das »Shelf Life Extension Program« aufgelegt (12, 13), um die Frequenz der Neubeschaffungen von etwa 120 Arzneimitteln zu senken. Dies ist möglich, da man den Gehalt der Arzneimittel bei der amerikanischen Food and Drug Administration (FDA) regelmäßig analysiert.
Ähnliche Untersuchungen finden auch bei der deutschen Bundeswehr statt. Ergebnisse stehen der Öffentlichkeit aber nicht zur Verfügung.
Um ein genaueres Bild zu erhalten, untersuchen wir am Institut für Biomedizinische und Pharmazeutische Forschung (IBMP), Nürnberg-Heroldsberg mithilfe der eigenen PEAKStab-Sammlung Arzneimittel, die bei Patienten über fünf Jahre hinaus gelagert wurden.
© Adobe Stock/ImageSine
Verlässt das Arzneimittel die Herstellerfirma in Richtung Großhändler und Apotheke, ist eine Lagerung bei ausreichend definierten Bedingungen garantiert. Nach Abgabe an den Patienten ist die korrekte Lagerung oft eher Zufall. Wie in den Medien oft kritisiert, heben viele Menschen ihre Arzneimittel im Badezimmer oder in der Küche auf, wo es zu warm und zu feucht sein kann. Oder die Medikamente werden in ein tropisches Urlaubsgebiet mitgenommen, wo sich beispielsweise ASS-Brausetabletten unter Extrem¬bedingungen von Feuchte und Temperatur schnell zersetzen. Idealerweise sollten Arzneimittel an einem kühlen, trockenen und dunklen Ort gelagert werden. Trotz einer möglicherweise unsachgemäßen Lagerung gilt das aufgedruckte Haltbarkeitsdatum.
Natürlich ist die Haltbarkeit auch abhängig von der Arzneiform und ob eine Verblisterung vorliegt. Daher sind Tabletten und Dragees in der Regel haltbarer als Salben, Gele, Cremes oder Flüssigkeiten. Letztere dürfen nach dem Öffnen der Tube oder der Flasche meist nur noch zeitlich begrenzt verwendet werden.
Auch hier sei einleitend festgestellt, dass der aktuelle Fall mit Paxlovid nicht dem von Tamiflu entspricht. Was tun, wenn ein Arzneimittel aufgrund einer Pandemie zur notfallmäßigen Behandlung von Patienten schnell zugelassen werden soll und zu diesem Zeitpunkt eine berechtigte Hoffnung auf Wirksamkeit besteht, aber keine ausreichenden Stabilitätsstudien vorliegen?
Genau dieser Fall ist 2022 mit Paxlovid eingetreten. Das von der Pharmafirma Pfizer entwickelte Paxlovid kam Anfang 2022 sowohl in Europa als auch in den USA zur Behandlung von Covid-19-Patienten auf den Markt (14). Zu diesem Zeitpunkt gab es die Impfstoffe verschiedener Hersteller, aber kein wirksames Mittel gegen die Covid-Erkrankung, das ambulant oral eingenommen werden und Klinikeinweisungen reduzieren konnte. Erste klinische Studien hatten ergeben, dass Paxlovid genau diese Anforderungen erfüllen könnte. Deshalb wurde es in einem »Fast-Track-Verfahren« ohne die üblichen Langzeitstabilitätsdaten zugelassen (Tabelle 2). Das Medikament besteht aus zwei Wirkstoffen/Tabletten:
Ritonavir ist seit fast 30 Jahren auf dem Markt und seine Stabilität gut untersucht. Die Substanz selbst ist chemisch stabil, liegt aber in zwei polymorphen Formen von unterschiedlicher Löslichkeit vor. Dabei kann das gut lösliche Polymorph in die schlechter lösliche Form »umkristallisieren«. Dies kann man heute durch eine entsprechende Formulierung vermeiden.
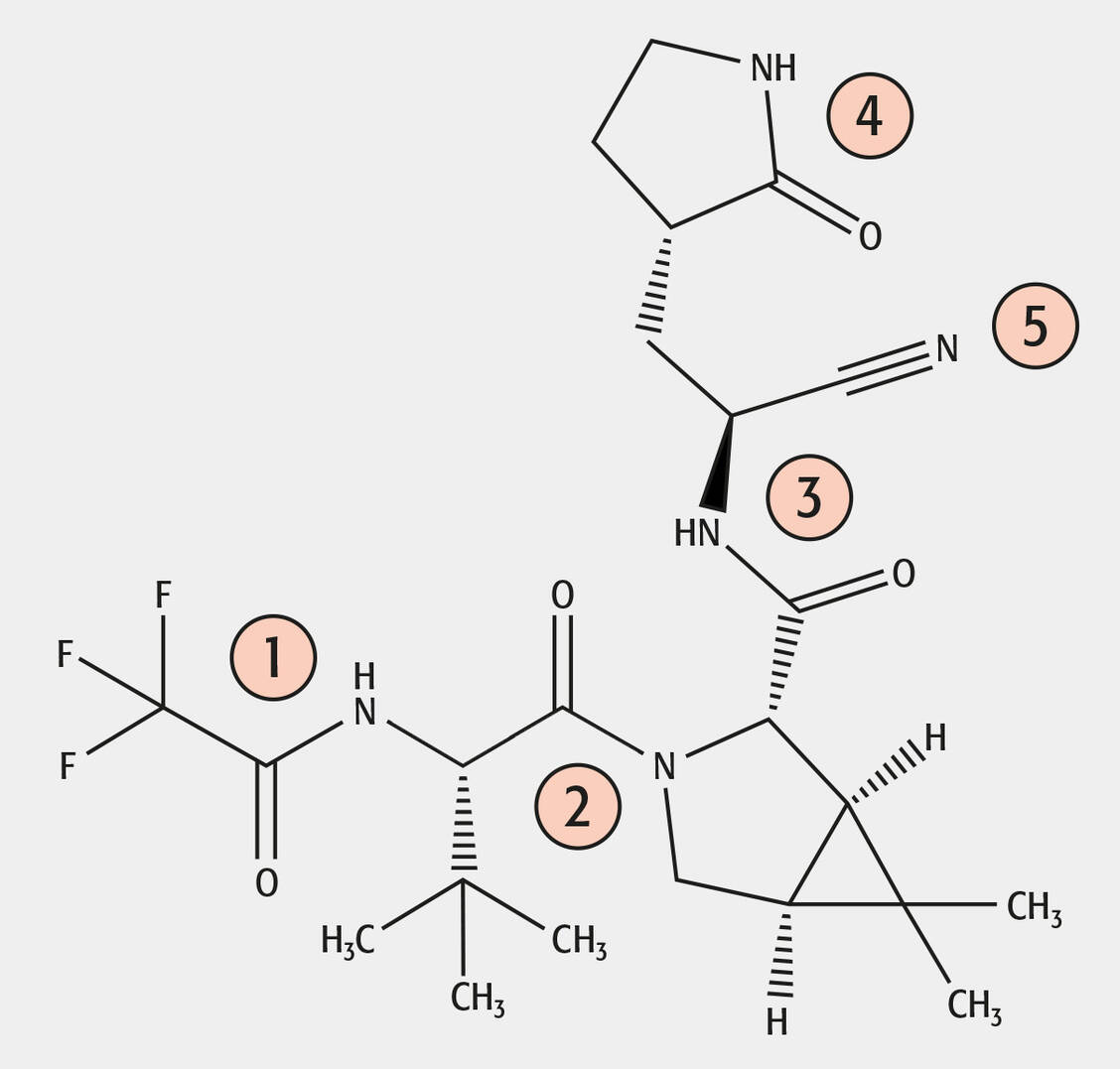
Abbildung: Strukturformel von Nirmatrelvir mit der Zuordnung der Amide und des Nitrils / © PZ/Holzgrabe
Zu Nirmatrelvir sind verständlicherweise bisher nur wenige Stabilitätsstudien bekannt. Das Molekül hat vier Amidfunktionen (Nr. 1–4 im Molekül) und eine Nitrilgruppe (Nr. 5), die hydrolytisch sowohl alkalisch als auch sauer gespalten werden können. Bemerkenswerterweise wird unter basischen Stressbedingungen nur das Nitril (Nr. 5) zu einem Amid hydrolysiert und am Amid 1 wird die Trifluoressigsäure abgespalten (Abbildung), wobei hier das entsprechende Amin entsteht (17, 18). Außerdem wurde eine Epimerisierung in Nachbarschaft zum Nitril (Nr. 5) beobachtet (19).
In Stressstudien mit Licht, Oxidation und Hitze erwies sich Nirmatrelvir als stabil (17, 20, 21). Bei jeweils zweistündiger Exposition von 1 M NaOH, 1 M HCl, 3 Prozent H₂O₂, Sonnenlicht und 80 °C wurde in einer neuen Studie nur im Alkalischen eine Zersetzung von knapp 30 Prozent beobachtet; alle anderen gefundenen Gehalte lagen über 96,6 Prozent (22).
| Datum | Ablauf der Zulassung |
|---|---|
| Jahr 2021 | |
| 27. April | Pfizer gibt die Entwicklung von Paxlovid™ bekannt und beginnt klinische Studien |
| 2. November | Preprint über erste klinische Daten |
| 5. November | erste Phase-II/III-Ergebnisse: weniger Krankenhausaufenthalte |
| 14. Dezember | endgültige Phase-III-Ergebnisse |
| 22. Dezember | Notzulassung durch FDA |
| Jahr 2022 | |
| 27. Januar | Zulassung durch EMA |
| 25. Mai | reguläre Zulassung durch FDA |
Aufgrund der 2022 fehlenden Stabilitätsdaten für Nirmatrelvir wurde Paxlovid mit einer Haltbarkeitsdauer von nur einem Jahr zugelassen, mit der Maßgabe, dass eine kontinuierliche Überprüfung der Stabilität erfolgen sollte, um gegebenenfalls das Verfallsdatum anzupassen.
Nachdem schon im November 2022 und im Januar 2023 die ersten Verfallsdaten überschritten waren, wurden die entsprechenden Batches untersucht. Dabei wurde kein Anzeichen von Zersetzung oder Gehaltsverlust festgestellt, sodass die Zulassungsbehörden in den USA, in Europa und Australien die Haltbarkeit um sechs auf 18 Monate verlängerten. Die Angaben auf den Verpackungen wurden nicht geändert, sondern man verwies auf einen BfArM-Link im Internet!
Dasselbe geschah im April und im Juli 2023, sodass nun eine Haltbarkeit von insgesamt 24 Monaten festgesetzt wurde. Somit konnten Packungen mit den Verfallsdaten zwischen 11/2022 und 12/2023 zwölf Monate weiter abgegeben werden. Allerdings durften Paxlovid-Packungen mit dem aufgedruckten Verfallsdatum 01/2024 nicht über dieses Datum hinaus abgegeben werden, da die verlängerte Haltbarkeit hier bereits berücksichtigt ist. Das bedeutet, dass Pfizer letztlich für Paxlovid eine Haltbarkeit von 24 Monaten festgeschrieben hatte. Aber warum?
Da die Haltbarkeit schon Anfang 2022 in der Schwebe war, hat das IBMP bereits im Oktober 2022 begonnen, den Gehalt von Nirmatrelvir-Tabletten mit unterschiedlichen Verfallsdaten in regelmäßigen Abständen mittels einer validierten RP-Hochleistungsflüssigkeitschromatografie mit massenspektrometrischer Detektion zu bestimmen. Die Messpunkte waren Oktober 2022, April 2023, April 2024 und November 2024 (Tabelle 3).
| Batch-Nummer | Verfallsdatum aufgedruckt | Verfallsdatum aktualisiert | Gehalt bestimmt am | Zahl untersuchter Tabletten | Wiederfindung in Prozent (SDV) |
|---|---|---|---|---|---|
| FX4624 | Januar 2023 | Juli 2023 | 04.10.2022 | 10 | 100,3 (3,6) |
| FT0016 | November 2022 | Mai 2023 | 18.04.2023 | 10 | 102,6 (4,1) |
| FT0016 | November 2022 | Mai 2023 | 04.04.2024 | 3 | 99,5 (3,2) |
| GN1533 | November 2023 | Februar 2024 | 04.04.2024 | 3 | 98,8 (3,7) |
| FT0016 | November 2022 | Mai 2023 | 12.11.2024 | 3 | 96,7 (3,4) |
Die erhobenen Daten kann man als »Real-World«-Daten betrachten, da die Packungen nicht mehr unter kontrollierten Bedingungen gelagert worden waren. Wie aus Tabelle 3 zu ersehen ist, liegen alle Gehalte, wie von der Q1A(2R)-Richtlinie [9] vorgegeben, zwischen 95 und 105 Prozent und unterscheiden sich nicht signifikant voneinander, auch wenn es einen kleinen Trend zu niedrigeren Gehalten gibt. Mit anderen Worten: Es hat keine wesentliche Veränderung stattgefunden, bei der »der Gehaltswert gegenüber dem Ausgangswert um mehr als 5 Prozent verringert« wäre (11). Dies entspricht auch den Beobachtungen mehrerer Stressstudien und insbesondere der von Yassin und Kollegen (22).
Da die FT-Chargen mit dem Verfallsdatum von 11/2022 bereits im Februar 2022 auf den Markt gekommen waren, mussten sie bei einem Verfall von einem Jahr spätestens im November 2021, eventuell auch früher, produziert worden sein. Das bedeutet, dass man von einer deutlich längeren Haltbarkeit als zwei Jahren ausgehen muss (23).

© Daniel Karmann
Umkehrphasen-Säule, Ascentis-Express RP-Amid, 50 mm × 4,6 mm, 2,7 µm; Gradienten mit den mobilen Phasen 0,1 Prozent wässrige Ameisensäure und 0,1 Prozent Ameisensäure in Acetonitril.
Die Quantifizierung von Nirmatrelvir erfolgte anhand des Peakflächenverhältnisses von Nirmatrelvir zu internem Standard. Die Linearität der Kalibrierungskurve in 0,1 Prozent Ameisensäure wurde von 0,610 bis 5,87 µg/ml nachgewiesen. Die untere Quantifizierungsgrenze wurde auf 0,610 µg/ml festgelegt. Es wurden keine Interferenzen für Nirmatrelvir und den internen Standard beobachtet.
Die Intra-Day-Präzision der Qualitätskontrollproben lag unter 6,1 Prozent mit einer analytischen Genauigkeit zwischen 99,0 Prozent und 102,8 Prozent. Die Inter-Day-Präzision der Qualitätskontrollproben lag unter 6,2 Prozent mit einer analytischen Genauigkeit zwischen 99,1 Prozent und 100,9 Prozent.
Die Bundesgesundheitsminister Jens Spahn und Karl Lauterbach haben bis 2022 in der Hochphase der Covid-19-Pandemie eine Million Packungen für die Patienten gekauft, die über Großhandel und Apotheken verteilt werden sollten. So sind auch andere Länder vorgegangen.
Zu Beginn 2024 waren in Deutschland noch etwa 400.000 Packungen gelagert, die jeweils für knapp 800 Euro von der Bundesrepublik eingekauft worden waren (24). Sie durften aufgrund einer Warnung von Pfizer aber nicht mehr (für 59,90 Euro) verteilt werden, da das Verfallsdatum angeblich überschritten war. Auch dies ist in vielen Ländern ähnlich. Das bedeutet, dass weltweit Millionen von Paxlovid-Packungen ohne wissenschaftlichen Beleg für eine Notwendigkeit vernichtet werden müssen.
Interessanterweise verkündete Pfizer kurz zuvor eine Preiserhöhung auf 1150 Euro (inklusive Mehrwertsteuer) pro Packung [25], die die Firma entsprechend den regulären arzneimittelrechtlichen Vertriebswegen seit 15. Januar 2024 in den Verkehr bringt. Dieser Abgabepreis muss von Patienten und Krankenkassen bezahlt werden. Es gibt nach Einschätzung der Autoren keinen Grund, ab 15. Januar 2024 von einem instabilen Medikament zu sprechen, sodass kommerzielle Interessen des Herstellers im Vordergrund gestanden haben könnten.
Es ist richtig, dass man die Stabilität eines Arzneistoffs und Arzneimittels detailliert untersuchen muss, um Patienten vor Arzneimitteln minderer Qualität zu schützen. Es gibt aber in der Literatur mehrere Untersuchungen, dass viele Arzneimittel weit über das Verfallsdatum hinaus noch immer die geforderte Qualität und damit Wirksamkeit haben.
Daher sollten die Zulassungsbehörden die regelhafte Festsetzung der Laufzeit von fünf Jahren überdenken, auch wenn die Hinweise für die Sinnhaftigkeit eines solchen Vorgehens »nur« aus beim Militär oder in der häuslichen Umgebung von Patienten gewonnenen Daten und nicht aus einem für Stabilität prädestinierten Labor stammen. In Europa dürften wohl nur angebrochene flüssige Arzneiformen ein Problem darstellen; dagegen sollten verblisterte angebrochene Arzneimittel problemlos weiter eingenommen werden können, wie unsere vielfältigen Studien zeigen.
Die Autoren versichern, dass sie weder von der Firma Pfizer noch einem anderen Hersteller Honorare oder Unterstützung für die hier zitierten Forschungsergebnisse erhalten haben.
Die getesteten Präparate entstammen der Sammlung PEAKStab des IBMP, Institut für Biomedizinische und Pharmazeutische Forschung, Nürnberg-Heroldsberg.
Angesichts möglicher Kostenersparnis, der vielfältigen Lieferengpässe von Arzneimitteln, aber auch der Nachhaltigkeit wäre die Verlängerung der Haltbarkeit nach entsprechenden Studien wohl berechtigt. Die dazu notwendigen Stabilitätsdaten müssten die Herstellerfirmen vorlegen, was aber offensichtlich von ihnen nicht unterstützt wird.
Der Fall Paxlovid zeigt nachdrücklich die Ablehnung weitergehender Stabilitätsuntersuchungen von Arzneimitteln über ihr Verfallsdatum hinaus durch die pharmazeutische Industrie, obwohl angenommen darf, dass solche beim Hersteller vorliegen und eine Selbstverständlichkeit sind. Umso wichtiger sind unabhängige Untersuchungen wie die am IBMP, die eine längere Haltbarkeit nahelegen.
Martina Kinzig studierte Chemie an der Universität Paderborn und wurde dort bei Professor Dr. Karl-Siegfried Boos promoviert. Anschließend arbeitete sie zunächst als Postdoc und seit 1993 als Laborleiterin am Institut für Biomedizinische und Pharmazeutische Forschung in Nürnberg-Heroldsberg. Sie war bei etwa 300 Methodenentwicklungen und ihren Anwendungen im Bereich der Klinischen Pharmakologie federführend. Das von ihr geleitete Labor ist GLP-zertifiziert. Eine Vielzahl ihrer Studien führten zu Arzneimittelzulassungen weltweit.
Ulrike Holzgrabe studierte Chemie und Pharmazie in Marburg und Kiel und habilitierte sich in Pharmazeutischer Chemie in Kiel. Nach mehrjähriger Professorentätigkeit in Bonn ist sie seit April 1999 als Professorin in Würzburg tätig, seit Mitte 2022 als Emerita. Professor Holzgrabe war von 2018 bis 2021 Vizepräsidentin der Universität Würzburg. In vielfältigen Positionen arbeitete sie am Deutschen und Europäischen Arzneibuch am BfArM und EDQM mit. Seit vielen Jahren forscht sie auf dem Gebiet der Antibiotika und der Analytik.
Fritz Sörgel studierte Pharmazie an der Goethe-Universität in Frankfurt/Main und wurde bei Professor Dr. Ernst Mutschler promoviert. Anschließend war er Postdoc bei Professor Dr. Leslie Benet an der School of Pharmacy, University of California, San Francisco. Es folgten eine Tätigkeit an der Rechtsmedizin in Erlangen sowie die Habilitation und Professur an der Universitätsklinik Essen. 1987 gründete Sörgel in Nürnberg das Institut für Biomedizinische und Pharmazeutische Forschung. Seither widmet er sich vielen Themen der Pharmakologie.
Das Virus SARS-CoV-2 hat unsere Welt verändert. Seit Ende 2019 verbreitet sich der Erreger von Covid-19 und stellt die Wissenschaft vor enorme Herausforderungen. Sie hat sie angenommen und rasch Tests und Impfungen, auch für Kinder, entwickelt. Eine Übersicht über unsere Berichterstattung finden Sie auf der Themenseite Coronavirus.


