 |  | Daniela Mackert |
 |
22.12.2023 16:30 Uhr |

Von seltenen Erkrankungen sind laut Definition nicht mehr als 5 von 10.000 Menschen in der Europäischen Union betroffen. Für diese ist die Krankheit massiv einschränkend sowie mitunter lebensbedrohlich und lebensverkürzend. / Foto: Getty Images/vchal
In der Europäischen Union gilt eine Erkrankung dann als selten, wenn nicht mehr als 5 von 10.000 Einwohnern davon betroffen sind (1). Das Kriterium für die Einstufung als seltene Erkrankung ist folglich die Prävalenz. Von den rund 30.000 bekannten Krankheiten werden ungefähr 8000 als selten eingestuft (1). Aktuellen Schätzungen zufolge betrifft das in Deutschland rund 4 Millionen Menschen und europaweit circa 30 Millionen (2, 3). Global und absolut betrachtet, kommen seltene Erkrankungen also gar nicht so selten vor. Dennoch sind mitunter weltweit nur ein paar Hundert Personen betroffen, weshalb seltene Erkrankungen lange vernachlässigt wurden; daher kommt auch der Name »Orphan Diseases«, also »verwaiste Krankheiten« (englisch: orphan = Waise).
Seltene Erkrankungen verlaufen oft chronisch und gehen meist mit gesundheitlichen Einschränkungen oder einer kürzeren Lebenserwartung einher. Nur die wenigsten sind heilbar oder lassen sich gut behandeln (1). Trotzdem gibt es bislang kaum kausale Therapieansätze oder wirksame symptomatische Behandlungsoptionen (4).
Orphan Diseases bilden ein breitgefächertes Spektrum meist sehr komplexer Krankheitsbilder. Dazu zählen sowohl gesamte Krankheitsgruppen, zum Beispiel Hämophilien, als auch onkologische Erkrankungen wie Hodgkin- und Non-Hodgkin-Lymphome, bestimmte Arten von Leukämien und verschiedene Ausprägungen des Pankreaskarzinoms. Weitere Beispiele sind bestimmte Erkrankungen des Herz-Kreislauf-Systems (Beispiel pulmonale Hypertonie), einige Stoffwechsel- und Autoimmunerkrankungen, Tuberkulose, Zytomegalievirus-Infektionen, systemischer Lupus erythematodes oder das Sjögren-Syndrom.
Circa 80 Prozent aller seltenen Erkrankungen – darunter beispielsweise Mukoviszidose, Hämophilien und Phenylketonurie – haben eine genetische Ursache. Etwa die Hälfte manifestiert sich bereits im Kindesalter, andere wiederum entwickeln sich erst im Erwachsenenalter (2).
Meist sind mehrere Organsysteme gleichzeitig betroffen und oftmals fehlen klare Leitsymptome. Da die Erkrankungen selten sind, gibt es nur wenige klinische Experten. Dies erschwert die medizinische Versorgung der Patienten und führt häufig zu Fehldiagnosen. Durchschnittlich wartet ein Betroffener sechs Jahre auf die korrekte Diagnose – ein langer, nervenaufreibender Weg für die Betroffenen (5).

Die meisten seltenen Erkrankungen – unter anderem die Mukoviszidose – haben eine genetische Ursache. / Foto: Getty Images/SDI Productions
Forschung und Studien werden durch fehlende demografische und epidemiologische Daten erschwert. Seltene Erkrankungen erfordern daher die Zusammenarbeit unterschiedlicher medizinischer Fachgruppen. Sogenannte Zentren für Seltene Erkrankungen (ZSE) bündeln Informationen zu spezialisierten Versorgungseinrichtungen. Für einen optimierten Informationsaustausch und eine bessere Versorgung der Betroffenen wurde 2010 das Nationale Aktionsbündnis für Menschen mit Seltenen Erkrankungen (NAMSE) gegründet, an dem unter anderem die Bundesministerien für Gesundheit sowie für Bildung und Forschung, ACHSE (Allianz Chronischer Seltener Erkrankungen) und 25 weitere Bündnispartner beteiligt sind (1).
Unter Orphan Drugs versteht man Arzneimittel zur Prävention, Diagnose und Behandlung von seltenen Erkrankungen (6). Deren Entwicklung stellt nach wie vor eine große Herausforderung dar – aus vielfältigen Gründen. Für eine erfolgreiche Arzneimittelforschung müssen genug Informationen zur Entstehung und den Krankheitsprozessen auf molekularer Ebene bekannt sein, was meistens nicht der Fall ist. Deshalb steht vor der Entwicklung eines Orphan Drugs eine aufwendige und mit hohen Kosten verbundene Grundlagenforschung. Dann müssen passende Behandlungsansätze abgeleitet und Wirkstoffe sowie Applikationsformen identifiziert und erprobt werden. Klinische Studien sind aufgrund der geringen Zahl an Betroffenen und deren räumlicher Verteilung mit großem Aufwand verbunden.
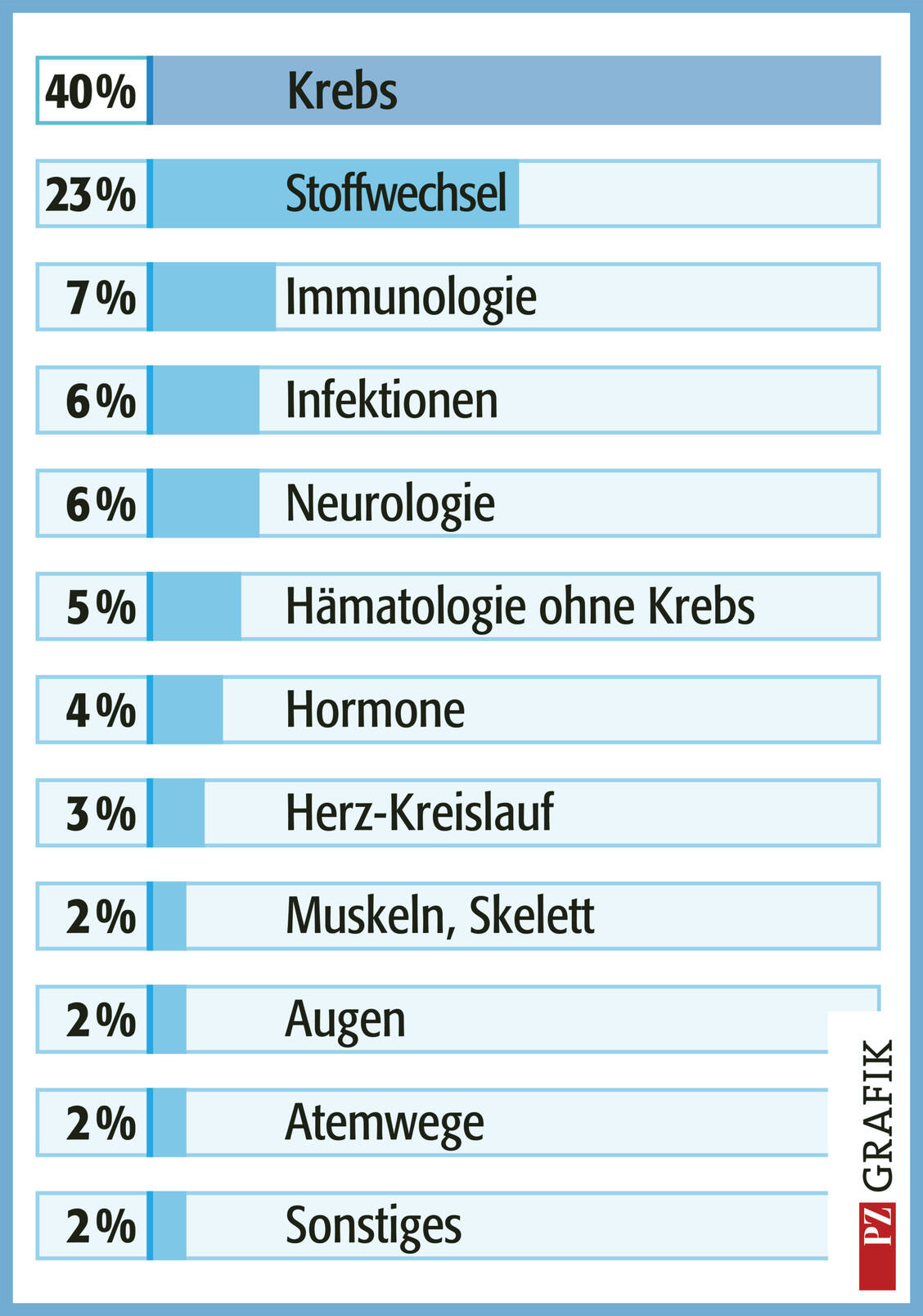
Grafik: Orphan-Drug-Zulassungen nach Indikationsgebieten und Erkrankungen (Stand Februar 2023). Aufgenommen wurden 218 Wirkstoffe (darunter 73, die keinen Orphan-Drug-Status mehr haben) gegen 171 Krankheiten. Modifiziert nach (8). / Foto: PZ/Stephan Spitzer
Die Notwendigkeit, verbesserte Rahmenbedingungen und Voraussetzungen zu schaffen, erkannten die USA bereits 1983 und verabschiedeten den Orphan Drug Act mit dem Ziel, die Erforschung, Entwicklung und Vermarktung dieser Arzneimittel zu fördern. 1991 folgte Singapur, 1993 Japan und 1998 Australien.
Schließlich wurden im Jahr 2000 auch für Europa die rechtlichen Grundlagen für Regelungen zu Orphan Drugs geschaffen und in der Verordnung (EG) Nr. 141/2000 beschrieben (7). Diese Verordnung regelt die Verfahren und Kriterien für die Ausweisung eines Arzneimittels als Orphan Drug durch die Europäische Kommission (»orphan designation«) (Kasten).
Auch wenn die Forschung weiterhin wirtschaftlich riskant ist, hat die Regelung zu einer deutlichen Zunahme der verfügbaren Orphan Drugs geführt: Seit Einführung der EG-Verordnung wurden mehr als 200 Orphan Drugs zugelassen. Aktuell sind in der EU etwa 150 Orphan Drugs (Stand Juli 2023) zugelassen. Hinzu kommen 73 weitere Medikamente, bei denen der Orphan-Drug-Status nach der Zulassung zurückgegeben wurde oder nach zehn Jahren abgelaufen ist (Grafik) (1). In den letzten Jahren machten Orphan Drugs durchschnittlich ein Drittel der jährlichen Neueinführungen von Medikamenten mit neuem Wirkstoff aus.
Eine vollständige Übersicht bietet die EU im »Community Register of orphan medicinal products«. Auch Orphanet stellt ein Verzeichnis der in Europa verfügbaren Orphan Drugs zur Verfügung (www.orpha.net/consor/cgi-bin/index.php).
In der Orphan-Drug-Verordnung bestimmt Artikel 3 die Kriterien, die ein Antragsteller wissenschaftlich nachweisen muss, damit sein Arzneimittel als Orphan Drug in der Europäischen Union anerkannt und ausgewiesen wird. Hierfür müssen mehrere Bedingungen erfüllt sein (Art. 3 VO (EG) Nr. 847/2000).
Von den insgesamt mehr als 200 verschiedenen onkologischen Erkrankungen erfüllt eine zunehmende Zahl den Status einer seltenen Krankheit. Dies liegt am fortschreitenden molekularen Wissen der Tumorbiologie und der verbesserten Diagnostik. Die Kenntnis der Subformen und speziellen genetischen oder molekularen Charakteristika von Tumoren hat eine immer stärkere Differenzierung ermöglicht. In der Folge werden immer mehr Orphan Drugs im onkologischen Sektor zugelassen und ermöglichen eine gezieltere Behandlung kleiner Patientengruppen.

Viele Orphan Drugs sind für onkologische Indikationen zugelassen. / Foto: Adobe Stock/Seventyfour
Ein besonderes Beispiel ist der monoklonale Antikörper Tremelimumab. Dieser erhielt von der Europäischen Arzneimittelagentur (EMA) gleich zwei Zulassungsempfehlungen: einmal als »Tremelimumab Astra-Zeneca« für die Indikation metastasiertes nicht kleinzelliges Lungenkarzinom (NSCLC) und zugleich als Orphan Drug unter dem Handelsnamen Imjudo® für die Orphan-Indikation hepatozelluläres Karzinom. Weitere Beispiele aus der Onkologie sind Tagraxofusp (Elzonris®) für den Einsatz bei blastisch plasmazytoiden dendritischen Zellneoplasien, woran EU-weit etwa 4500 Personen erkrankt sind, sowie Niraparib (Zejula®) für die Indikation Ovarialkarzinom, das laut Statistik ungefähr 190.000 Frauen in der EU betrifft.
Für eine sehr kleine Patientenpopulation (etwa 450 Betroffene EU-weit) wurde Asfotase alfa (Strensiq®) zur Behandlung der Hypophosphatasie, einer genetisch bedingten, potenziell tödlich verlaufenden Erkrankung des Knochenstoffwechsels, zugelassen. Der Wirkstoff Macitentan (Opsumit®) dient zur Behandlung von pulmonaler Hypertonie, die ebenfalls als Orphan Disease eingestuft wird und etwa 79.000 Personen in der EU betrifft. Allein diese Beispiele verdeutlichen die Bandbreite der Betroffenenpopulationen bei Orphan Diseases.
Um die Entwicklung und Vermarktung von Orphan Drugs attraktiv zu gestalten, hat die Europäische Union spezielle Anreize für pharmazeutische Unternehmen geschaffen. Damit wird Herstellern auch in kleinen Märkten die Aussicht gegeben, die Kosten von Forschung und Entwicklung, Produktion und Vermarktung zu decken und einen Gewinn zu erzielen (8, 9). Hierzu zählen:
Arzneimittel für seltene Erkrankungen durchlaufen bei der EMA ein zweistufiges Verfahren. Dem Ausschuss für Orphan Medicinal Products (COMP: Committee for Orphan Medicinal Products) obliegt vor allem die Prüfung von Anträgen auf Ausweisung als Orphan Drug. Stimmt die Europäische Kommission dem Votum des COMP zu, wird das Produkt in das Orphan-Drug-Register der EU aufgenommen.

Für die pharmazeutische Industrie gibt es zahlreiche Anreize, auf dem Gebiet der seltenen Erkrankungen zu forschen. / Foto: Getty Images/Morsa Images
Darüber hinaus muss das Medikament zusätzlich in Deutschland noch das nationale AMNOG-Verfahren zur Bewertung des Zusatznutzens durchlaufen. Dabei gilt nach § 35a Absatz 1 SGBV der medizinische Zusatznutzen (»significant benefit«) bereits durch die europäische Zulassung als belegt.
Nach erfolgter Zulassung überprüft die EMA am Ende des fünften Jahres im Markt, ob die Orphan-Kriterien weiterhin erfüllt werden. Die Marktexklusivität erlischt, wenn das Präparat eines anderen Anbieters sicherer, wirksamer oder anderweitig therapeutisch überlegen ist. Sobald das Orphan-Medikament einen Jahresbruttoumsatz von 30 Millionen Euro übersteigt, wird es rechtlich wie ein gewöhnliches Arzneimittel behandelt. Dann muss ein normales Zusatznutzendossier beim G-BA eingereicht werden und es erfolgt eine Nutzenbewertung durch das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) mit anschließenden Verhandlungen über den Erstattungsbetrag.
Trotz der unbestreitbaren Vorteile von Orphan Drugs für die Patienten gibt es immer wieder kontroverse Diskussionen um einzelne Aspekte dieser Wirkstoffe (10–15). Folgende Fragen werden häufig zu Orphan Drugs gestellt.
Bei allen Arzneimitteln müssen die pharmazeutischen Unternehmer im Rahmen des Zulassungsprozesses Wirksamkeit, Verträglichkeit und pharmazeutische Qualität belegen. Doch die üblichen randomisierten, kontrollierten klinischen Studien mit Hunderten oder Tausenden Patienten sind bei seltenen Erkrankungen kaum oder nicht möglich. Auch spezielle statistische Methoden bei der Auswertung der Studien sind nur begrenzt anwendbar.
Für diese Fälle haben die Experten der EMA eine Leitlinie erstellt, in der Alternativen dargelegt werden. Dazu müssen zunächst geeignete Modelle zur Prüfung der Arzneimittelwirkungen, Prüfkriterien für die Erfolgsmessung, zum Beispiel Verbesserung der Lebensqualität oder Zeit bis zum Fortschreiten der Erkrankung, und statistische Methoden zur Auswertung der Studienergebnisse aufgezeigt werden.
Es kann vorkommen, dass die Durchführung einer klinischen Studie nicht möglich ist. In diesen Ausnahmefällen kann eine Zulassung auch auf Basis sehr gut dokumentierter Berichte über einzelne Behandlungsfälle erteilt werden. Dennoch müssen weitere Belege für die Wirksamkeit und Unbedenklichkeit des Präparates erbracht werden.
Im Anschluss durchläuft das Orphan Drug das Zulassungsverfahren. Dieses selbst ist nicht nennenswert schneller, denn Orphan Drugs unterliegen denselben Anforderungen an die klinische Erprobung und Zulassung wie alle anderen Medikamente.
Der Zusatznutzen von Orphan Drugs gerät immer wieder in die Kritik, da er aufgrund der speziellen regulatorischen Rahmenbedingungen bestimmte Herausforderungen und ethische Fragen aufwirft. Aufgrund niedriger Fallzahlen sind Studien mit deutlich geringeren Teilnehmermengen erlaubt und der Nutzen muss nicht gegenüber einer zweckmäßigen Vergleichstherapie belegt werden.
Die Evidenz für den Zusatznutzen kann der Gemeinsame Bundesausschuss (G-BA) nach Markteinführung meist nur anhand der Zulassungsunterlagen des Herstellers ermitteln. Ist keine eindeutige Nutzenbewertung (nicht quantifizierbarer Zusatznutzen) möglich, wird einem Orphan Drug ein »fiktiver Zusatznutzen« bescheinigt. Mit der Zulassung gilt damit der Zusatznutzen für Orphan Drugs als belegt.
In einer Analyse hat das IQWiG 41 Orphan-Drug-Bewertungen identifiziert, für die seit 2011 sowohl eine spezielle Orphan-Bewertung als auch eine nachfolgende reguläre Nutzenbewertung erfolgte. Betrachtet wurden 20Wirkstoffe, denn einige Stoffe waren für mehrere Anwendungsgebiete zugelassen. Bei 22 der 41 Bewertungen (54 Prozent) konnte in der regulären Nutzenbewertung kein Zusatznutzen (»nicht belegt«) festgestellt werden. Zudem ergab die Analyse, dass eine reguläre Bewertung des Zusatznutzens in der Regel erst nach mehreren Jahren erfolgte (teilweise bis zu neun Jahre; im Mittel drei Jahre). Bei Orphan Drugs, die die Umsatzschwelle von (der bis Ende 2022 noch gültigen) 50 Millionen Euro jährlich nicht erreichten, fand gar keine reguläre Nutzenbewertung statt. Thomas Kaiser, Leiter des IQWiG, mahnte an, dass die Qualität der Patientenversorgung darunter leiden könne und neue Arzneimittel möglicherweise ohne Datengrundlage bevorzugt eingesetzt würden (14).
Diese Analyse bezweifelt keineswegs den Nutzen und die Notwendigkeit von Orphan Drugs, sondern zielt darauf ab, die Bewertungsverfahren und Transparenz in Bezug auf den Nutzen zu verbessern, damit die Ressourcen sinnvoll eingesetzt werden und die Patienten den größtmöglichen Nutzen haben. Dies erfordert eine fortlaufende Diskussion und Anpassung der regulatorischen Rahmenbedingungen und Bewertungsmethoden.
Seit Einführung der EG-Verordnung wurden in Europa mehr als 200 Orphan Drugs zugelassen. Entsprechend dynamisch stiegen die Ausgaben für die Krankenkassen. Jürgen Windeler, ehemaliger Leiter des IQWiG, befürchtet, dass Orphan Drugs wesentliche Kostentreiber in der GKV darstellen (14).

Mit fortschreitendem molekularen Wissen in der Tumorbiologie und einer verbesserten Diagnostik werden immer mehr spezielle genetische oder molekulare Subformen von Tumoren erkannt. Diese werden mitunter als Orphan Disease eingestuft. / Foto: Getty Images/Andrew Brookes
Tatsächlich stieg der Anteil von Orphan Drugs an den gesamten Nettokosten für Arzneimittel von 2011 bis 2020 von 4 auf 11,8 Prozent am gesamten GKV-Markt an und entsprach im Jahr 2021 etwa 6,8 Milliarden Euro. Hiervon entfallen mit 3,5 Milliarden Euro rund 52 Prozent der Nettokosten der Orphan-Arzneimittel in Deutschland auf Krebserkrankungen.
So komplex sich oft das Krankheitsbild darstellt, so komplex sind auch die Entwicklung und Produktion solcher Medikamente, was sich auf die Endkosten auswirkt. Die Kosten pro Patient in einer klinischen Studie für eine seltene Erkrankung sind oft deutlich höher als bei häufigeren Erkrankungen.
Unter »Slicing« oder auch »Orphanisierung« versteht man die Strategie, dass Pharmaunternehmen das Orphan-Drug-Regelwerk samt den exklusiven Marktrechten und anderen Vorteilen für ihre Medikamente ausnutzen könnten, indem sie aus größeren Anwendungsgebieten Untergruppen »herausschneiden« (»Slicing«). Die Annahme ist, dass quasi künstlich kleine Subpopulationen entstehen, bis die Zahl der zu behandelnden Patienten der einer Orphan Disease entspricht. Ein solches Vorgehen erlaubt die EMA jedoch nicht und es wird in der Regelung (EMA/COMP/15893/2009) explizit verboten.
Man spricht von »Trojanern«, wenn Arzneimittel ursprünglich als Orphan Drug entwickelt wurden, um von den regulatorischen und wirtschaftlichen Vorteilen zu profitieren, und später – unter Beibehaltung der Orphan-Vorteile – für eine häufigere Erkrankung zur Zulassung gebracht werden.
Dies ist jedoch ein Irrtum, da ein Arzneimittel seinen Orphan-Drug-Status verliert, sobald mehr als 5 von 10.000 Einwohnern daran leiden oder es zusätzlich für eine Nicht-Orphan-Anwendung zugelassen wird. Allerdings kann der gleiche Wirkstoff sowohl in einem Orphan Drug als auch einem anderen Medikament eingesetzt werden; ein aktuelles Beispiel ist Tremelimumab. Das ist jedoch nur möglich, wenn es sich um verschiedene Indikationen handelt und Entwicklung, Zulassung und Vermarktung strikt getrennt ablaufen.

Der »Rare Disease Day« ist eine weltweite Bewegung: Seit 2008 vereinen sich am letzten Februartag – dem 28. oder 29 – Menschen auf der ganzen Welt, um auf die Anliegen und Bedarfe von Menschen mit seltenen Erkrankungen aufmerksam zu machen. / Foto: Adobe Stock/Rana
Auch wenn in den vergangenen Jahren deutliche Fortschritte bei der Entwicklung und Zulassung von Orphan Drugs erzielt wurden, so sind bislang nur für etwa 2 Prozent der seltenen Erkrankungen Orphan Drugs zugelassen. In Deutschland profitieren Betroffene in besonderem Maß, was nicht zuletzt eine Folge der deutschen Orphan-Drug-Regelung im AMNOG ist. In der Regel übernehmen die Krankenkassen die Therapiekosten.
In Europa ist die Kostenübernahme der teuren Arzneimittel jedoch kein Standard. Daher möchte die Europäische Kommission solidarischer agieren und will eine neue Arzneimittelstrategie entwickeln, die Orphan Drugs füralle EU-Bürger zugänglich und erschwinglich machen soll. Dies verlangt nach adäquaten Preisgestaltungs- und Kostenerstattungsregularien in den Mitgliedstaaten.
Fakt ist, dass die Orphan Drug-Verordnung und die damit verbundene Strategie erfolgreich sind und die notwendige Forschung und Entwicklung bestärkt haben. Dennoch ist zum Wohl der Patienten eine stetige Verbesserung des Regelungsumfelds notwendig. Letztlich kommt es auf die richtige Balance an.
Daniela Mackert studierte Pharmazie an der Julius-Maximilian-Universität Würzburg, war dort als wissenschaftliche Mitarbeiterin und von 2006 bis 2013 in öffentlichen Apotheken tätig. Von 2014 bis 2018 war sie Chefredakteurin »Schulmedizin« bei der Mediengruppe Oberfranken und absolvierte berufsbegleitend den Masterstudiengang »Master of Health Business Administration (MHBA)« am Lehrstuhl für Gesundheitsmanagement an der Friedrich-Alexander-Universität Erlangen-Nürnberg. Sie übernahm die Teamleitung »Content Marketing« der Mediengruppe Oberfranken, bevor sie sich 2020 als freiberufliche Medizinjournalistin selbstständig machte.


