 |  | Katharina Holl |
 |
28.11.2021 08:00 Uhr |
Während im akuten Schub der Multiplen Sklerose nach wie vor Glucocorticoide hochdosiert gegeben werden, gibt es in der immunmodulatorischen Dauertherapie neue Therapiestrategien. / Foto: Adobe Stock/LStockStudio
Der Erkrankungsgipfel der MS liegt zwischen dem 20. und 40. Lebensjahr. Sie stellt somit die häufigste neurologische Erkrankung junger Erwachsener dar. Aber auch Kinder und ältere Menschen können erkranken (1). In den letzten Jahren hat die Zahl der Diagnosen in Deutschland auf derzeit 18 pro 100.000 Versicherte zugenommen (2). Es ist allerdings unklar, ob hierfür tatsächlich eine Zunahme der Erkrankungshäufigkeit oder die verbesserte Diagnostik verantwortlich ist.
Die MS ist autoimmun vermittelt. Eine besondere Rolle spielen zunächst periphere Lymphozyten, die Strukturen des ZNS angreifen. Dies führt zu multiplen Läsionen, die die Symptome und später bleibende Behinderungen auslösen. In späteren Phasen setzen sich entzündliche Prozesse im ZNS selbst weiter fort (3). Zu den häufigsten Symptomen eines Schubs zählen Sehstörungen auf einem Auge aufgrund einer Entzündung des Sehnervs (Optikusneuritis), Taubheitsgefühl und/oder Kribbeln in einem Arm oder Bein, Gang- oder Gleichgewichtsstörungen sowie Störungen der Blasen- oder Darmentleerung.
Man unterscheidet verschiedene Verlaufsformen (4):
Wenn die Diagnose einer MS bei der ersten klinischen Manifestation (noch) nicht möglich ist, weil beim Patienten in der Magnetresonanztomografie (MRT, Kernspintomografie) bisher keine räumliche und zeitliche Dissemination (siehe unten) nachweisbar ist, spricht man vom klinisch isolierten Syndrom (KIS) (5).

Vorrangiges Therapieziel ist es, das Auftreten von Schüben und das Fortschreiten der Behinderung aufzuhalten. / Foto: Adobe Stock/Adam Wasilewski
Aufgrund der umfangreichen Forschungsergebnisse der letzten Jahre wurde eine Aktualisierung der Leitlinie von 2012 erforderlich. Im Februar 2021 wurde die S2k-Leitlinie »Diagnose und Therapie der Multiplen Sklerose, Neuromyelitis-Optica-Spektrum- und MOG-IgG-assoziierte Erkrankungen« veröffentlicht (1). Die weitere Aktualisierung soll von nun an mindestens jährlich erfolgen, falls erforderlich auch früher (Living Guideline). Erstmals haben auch Betroffene und Patientenvertreter an der Leitlinie mitgearbeitet, um deren Bedürfnissen verstärkt Rechnung zu tragen. Welches sind die wichtigsten Neuerungen hinsichtlich Diagnostik und Therapie?
In der Diagnostik gelten die sogenannten McDonald-Kriterien für die Magnetresonanztomografie, die 2017 zuletzt aktualisiert wurden (6). Diese Kriterien besagen: Für die sichere Diagnose der RRMS muss je nach Anzahl der bisherigen Schübe und der klinischen Symptome eine räumliche und/oder zeitliche Dissemination (Aus-/Verbreitung) von ZNS-Läsionen nachweisbar sein (Tabelle 1). Dabei bedeutet räumliche Dissemination (Dissemination in Space, DIS) das Vorhandensein mindestens einer MS-typischen Läsion der weißen Hirnsubstanz in mindestens zwei von vier definierten Hirnregionen. Die zeitliche Komponente (Dissemination in Time, DIT) umfasst entweder den gleichzeitigen Nachweis kontrastmittelanreichernder und nicht-kontrastmittelanreichernder Läsionen oder aber den Nachweis einer neuen Läsion (neben alten) in einem Folge-MRT.
| Zahl der Schübe | Objektivierbare klinische Manifestationen | Erforderliche diagnostische MRT-Kriterien |
|---|---|---|
| ≥ 2 | ≥ 2 | keine |
| ≥ 2 | 1 | DIS: weiterer symptomatischer Schuboder DIS-MRT |
| 1 | ≥ 2 | DIT: weiterer Schub oderDIT-MRT odercharakteristischer Liquorbefund |
| 1 | 1 | DIS und DIT |
Das bedeutet im Umkehrschluss, dass der Nachweis einer akuten entzündlichen Aktivität mittlerweile nicht mehr zwingend erforderlich ist, wenn sich bereits aus der klinischen Symptomatik und/oder dem Nachweis älterer Läsionen im MRT ausreichende Hinweise auf eine zurückliegende Entzündung ergeben. Folglich kann die Diagnose RRMS oftmals früher gestellt werden als noch vor einigen Jahren.
Kriterien für die Diagnose einer PPMS sind eine klinische Progression über mindestens ein Jahr in Verbindung mit zwei der folgenden Kriterien:
Zu beachten ist, dass die im MRT nachweisbaren Läsionen zwar typisch, aber nicht vollständig MS-spezifisch sind. Um Fehldiagnosen auszuschließen, ist daher auch der Ausschluss von Differenzialdiagnosen, unter anderem virale oder bakterielle Infektionen, andere demyelinisierende, vaskuläre oder rheumatologische Erkrankungen, Enzephalitiden und Tumoren, erforderlich (7).
Die Standarddiagnostik bei MS umfasst daher ein kraniales und spinales MRT, Liquordiagnostik sowie Borrelien- und Lues-Serologie und gegebenenfalls intrathekale Antikörper gegen Masern-, Röteln- und Varicella-zoster-Viren. Zur Differenzialdiagnostik werden oft auch elektrophysiologische Untersuchungsmethoden (visuell evozierte Potenziale) eingesetzt (1).
Als MS-Schub definiert man das Neuauftreten oder die Reaktivierung neurologischer Defizite (Missempfindungen wie Kribbeln oder Taubheit, Sehstörungen, Probleme mit der Darm- oder Blasenfunktion, Gang- oder Gleichgewichtsstörungen), die mindestens 24 Stunden anhalten, mehr als 30 Tage nach einem vorhergehenden Schub auftreten und nicht durch anderweitige Ursachen bedingt sind (7).
Bezüglich der Schubtherapie gibt es in der aktuellen Leitlinie keine Neuerungen. Standard ist die möglichst baldige Therapie mit hoch dosierten (500 bis 1000 mg/Tag) Glucocorticoiden, meist Methylprednisolon, über drei bis fünf Tage (1). In einem Cochrane-Report wurde eine günstige Wirkung auf die Schubsymptome dargelegt; dagegen gibt es keine Evidenz für das Verhindern neuer Schübe oder eine Abnahme der Langzeitbehinderung (9). Obwohl kein Beleg für einen Wirkunterschied zwischen oraler und intravenöser Gabe existiert (10), wird die intravenöse Gabe aus Praktikabilitätsgründen (orale Darreichungsformen gibt es nur bis zu einer Dosis von 40 mg) bevorzugt.

Im akuten Schub werden Glucocorticoide hoch dosiert gegeben, bevorzugt als Infusion. / Foto: Adobe Stock/ISO K Medien GmbH
Die Entscheidung für eine Schubtherapie trifft der behandelnde Arzt je nach Schwere des Schubs, Wirksamkeit und Verträglichkeit früherer Schubtherapien, Komorbiditäten und Kontraindikationen. Die Nebenwirkungen dieser kurzzeitigen Therapie sind begrenzt. Um Schlafstörungen zu vermeiden, sollten die Patienten die Infusion morgens erhalten.
Bei unzureichendem Effekt folgt eine Eskalation mit hochdosierten Corticosteroiden bis zu 2000 mg/Tag über drei bis fünf Tage. Alternativ oder zusätzlich kommen eine Plasmapherese oder Immunadsorption (selektive Entfernung von Immunglobulinen aus dem Plasma) zum Einsatz. Diese kann auch schon zu Beginn des Schubs erfolgen, vor allem wenn eine Glucocorticoid-Therapie nicht möglich ist und/oder das Verfahren bereits vorher gut wirksam war. Evidenz aus kontrollierten Studien ist allerdings rar (1).
Die Schwierigkeit bei der Therapie der MS besteht darin, dass der Krankheitsverlauf sehr variabel ausfällt und sich kaum vorhersagen lässt. Gewisse Anhaltspunkte bilden die Zahl und Schwere der bisherigen Schübe, Entzündungsaktivität und Läsionslast sowie der Liquorbefund.
Ein Grundsatz lautet deshalb, dass der Arzt die Therapie gemeinsam mit dem Patienten abhängig von der Aktivität der Erkrankung und unter Berücksichtigung der möglichen Nebenwirkungen und Kontraindikationen auswählt. Therapieziel ist es, klinische Symptome, das heißt Schübe und Krankheitsprogression zu verhindern oder zumindest zu reduzieren.
Auch für das Apothekenteam ist es wichtig, dem Patienten zu vermitteln, dass die Erkrankung nicht heilbar, aber durch frühzeitige und konsequente Therapie in ihrem Verlauf günstig beeinflussbar ist.
Grundsätzlich kann der Patient mit einer Immuntherapie bereits nach dem ersten Schub beginnen, auch dann, wenn nur ein klinisch isoliertes Syndrom gesichert ist. Bei einem isolierten Schub, der nicht als KIS klassifiziert werden kann, zum Beispiel einer isolierten Optikusneuritis oder isolierten Myelitis, ist ein Therapiebeginn dagegen nur in Ausnahmefällen angezeigt. Geht der Arzt aufgrund einer nur geringen Krankheitsaktivität von einem langsamen Verlauf aus, ist unter engmaschiger Kontrolle auch bei gesicherter RRMS ein Zuwarten ohne Immuntherapie möglich.
Für das sofortige Einleiten einer Therapie direkt nach dem ersten Schub sprechen dagegen:
In der bisherigen Version der Leitlinie (11) gab es ein Stufenschema zur Therapie der RRMS. Neu ist, dass die Therapeutika in der aktuellen Leitlinie nun anhand der Reduktion der Schubrate in drei Wirksamkeitskategorien eingeteilt werden (Tabelle 2). Wichtig: Diese Einteilung basiert nicht auf Studien, sondern auf Erfahrungswerten. Weiterhin ist praxisnah beschrieben, wann die Therapie begonnen oder eskaliert und wann sie beendet werden sollte.
| Kategorie | Relative Reduktion der Schubrate (verglichen mit Placebo) | Wirkstoffe |
|---|---|---|
| 1 | 30 bis 50 Prozent | Beta-Interferon, PEG-InterferonDimethylfumarat, GlatirameroideTeriflunomid |
| 2 | 50 bis 60 Prozent | CladribinS1P-Rezeptor-Modulatoren: Fingolimod, Ozanimod, Ponesimod |
| 3 | > 60 Prozent oder> 40 Prozent verglichen mit Kategorie 1 | AlemtuzumabCD20-Antikörper: Ocrelizumab, Rituximab (off Label)Natalizumab |
Mitoxantron sowie Azathioprin sollten wegen der schlechten Studienlage nur noch als Reservearzneistoffe, intravenös verabreichte Immunglobuline überhaupt nicht mehr verwendet werden.
Schwere Nebenwirkungen sind in den beiden höheren Kategorien tendenziell häufiger. Für die alltägliche Verträglichkeit insgesamt trifft das jedoch nicht grundsätzlich zu; diese kann bei Therapeutika der Kategorie 1 durchaus schlechter sein. Bei der Wahl eines konkreten Arzneistoffs spielen vorrangig das Nebenwirkungsprofil sowie die Patientenpräferenz eine Rolle.
Zu Beginn verordnet der Arzt normalerweise Arzneimittel der Kategorie 1, zum Beispiel Beta-Interferone (IFN-β), Dimethylfumarat (DMF) oder Glatirameracetat, es sei denn, ein hochaktiver Verlauf ist wahrscheinlich. Bei Patientinnen mit Kinderwunsch sollten DMF und Teriflunomid aufgrund der Teratogenität möglichst vermieden werden.
Treten innerhalb der nächsten zwei Jahre trotz Therapie ein weiterer Schub oder weitere Läsionen im MRT auf, sollte man auf Wirkstoffe der Kategorie 2 oder 3 wechseln. Dies sind in Kategorie 2 zum Beispiel Cladribin und Sphingosin-1-Phosphat-(S1P)-Rezeptor-Modulatoren wie Fingolimod, Ozanimod oder Ponesimod. Zur Gruppe 3 gehören zum Beispiel Natalizumab und CD20-Antikörper wie Ocrelizumab. Ein Wechsel auf andere Wirkstoffe der Kategorie 1 oder eine höhere Dosierung wird dagegen nur empfohlen, wenn patientenindividuelle Gründe gegen einen Umstieg in eine höhere Wirksamkeitskategorie sprechen (1). Der Therapiealgorithmus für die Ersteinstellung und Eskalation ist in Abbildung 1 dargestellt.
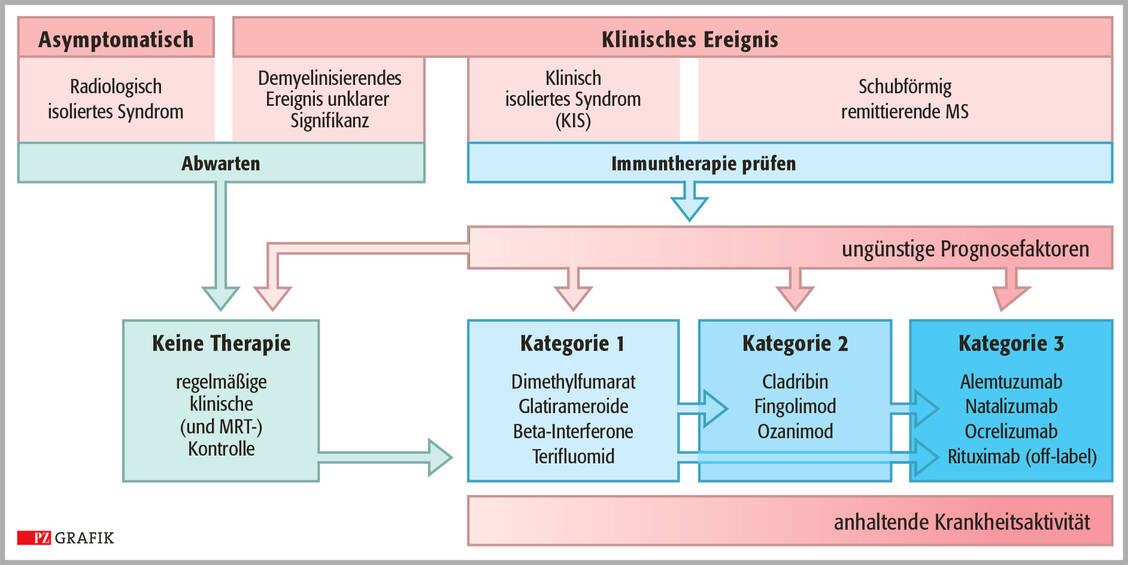
Abbildung 1: Therapiealgorithmus bei Ersteinstellung sowie Therapieeskalation bei Patienten mit RRMS; modifiziert nach (1) / Foto: PZ/Stephan Spitzer
Bisher gibt es leider keine eindeutigen Kriterien, einen potenziell aggressiven Krankheitsverlauf festzustellen (12, 13). Die Leitlinienautoren schlagen für therapienaive Patienten drei Kriterien vor, die vermutlich einen schweren Verlauf prognostizieren:
Diesen Patienten sollten direkt Wirkstoffe der Kategorien 2 oder 3 angeboten werden. Die Kategorie 3 sollte bei hoher Schubaktivität und hoher Läsionslast, polysymptomatischen Schüben sowie inkompletter Rückbildung in Betracht gezogen werden.
Zur Verlaufskontrolle dienen regelmäßige MRT-Kontrollen (anfangs zum Beispiel alle drei bis sechs Monate). Später hängt die Frequenz vom weiteren Krankheitsverlauf ab (1).
Insgesamt am häufigsten kommen Arzneistoffe der Kategorie 1 zum Einsatz. Wie auch viele Therapeutika der höheren Kategorien werden die meisten davon parenteral verabreicht. Der Apotheker kann die Injektionstechnik demonstrieren, idealerweise initial und danach in regelmäßigen Abständen.
Unter Interferonen und Glatiramer treten als Nebenwirkungen sehr häufig Reaktionen an der Injektionsstelle, zum Beispiel Rötung und Schwellung, sowie bei Interferonen auch grippeähnliche Symptome wie leichtes Fieber, Kopf- und Gelenkschmerzen und Schüttelfrost auf. Hier ist der Hinweis sehr hilfreich, dass diese meist im Verlauf der Behandlung nachlassen. Die Patienten können beispielsweise Paracetamol und Ibuprofen zur Linderung grippeähnlicher Symptome einnehmen.
DMF kann zu einer Depletion weißer Blutzellen führen, weshalb die Patienten dazu ermutigt werden sollen, unter der Therapie regelmäßig (alle sechs Wochen im ersten Jahr, danach alle drei bis sechs Monate) Blutbildkontrollen vornehmen zu lassen. Bei den meisten Menschen tritt allerdings kein relevanter Effekt auf die Immunabwehr auf. Weiterhin treten unter DMF und auch Teriflunomid oft Magen-Darm-Beschwerden auf, die aber meist im Verlauf der Therapie nachlassen.
Bei allen Arzneistoffen aus der Kategorie 2 sind Aspekte der Familienplanung und Verhütung zu beachten (siehe Beitrag »Chronisch Krank: Verhütung und Kinderwunsch«). Cladribin weist eine sehr lange Wirkdauer auf, was hinsichtlich der langen Dosierintervalle vorteilhaft, wegen fehlender Reversibilität bei eintretender Schwangerschaft jedoch nachteilig ist. S1P-Modulatoren (Fingolimod, Ozanimod, Ponesimod) haben dagegen eine kurze Halbwertszeit. Als besonderer Aspekt ist hier allerdings der relativ starke Rebound-Effekt (erneutes Auftreten von Schüben) zu nennen. S1P-Modulatoren verringern die Pulsfrequenz und die Erregungsleitung am Herzen. Hier kann das Apothekenteam auf mögliche Begleitmedikationen achten, die diese Effekte verstärken können.

Viele MS-Therapeutika erfordern eine sichere Verhütung. Mitunter reichen hormonelle Kontrazeptiva alleine nicht aus und es müssen Methoden kombiniert werden. / Foto: Adobe Stock/nenetus
Bei den Wirkstoffen der Kategorie 3 ist insbesondere das Auftreten einer progressiven multifokalen Enzephalopathie (PML) unter Natalizumab relevant. Daher sollte vor Therapiebeginn der JC-Virus(JCV)-Antikörperstatus bestimmt werden (1). Bei 10 Prozent der seronegativen Personen tritt jedoch im Verlauf eine Serokonversion auf (14). Damit steigt das Risiko für eine PML stark an, sodass der Antikörperstatus alle sechs Monate bestimmt werden sollte. Bei einer eindeutigen Serokonversion sollte der Arzt den Patienten zeitnah auf ein anderes Therapeutikum (Ocrelizumab, off Label Rituximab) umstellen. Hierbei ist anzumerken, dass Rituximab nicht für die Indikation MS zugelassen ist, aber von den Leitlinienautoren dennoch gleichwertig mit Ocrelizumab genannt wird.
Unter Alemtuzumab können noch bis zu vier Jahre nach Therapieende zerebrovaskuläre Nebenwirkungen, sekundäre Autoimmunerkrankungen und opportunistische Infektionen auftreten. Daher sollte man es nur noch einsetzen, wenn andere Wirkstoffe nicht infrage kommen (1).
Nach aktueller Studienlage sind CD20-Antikörper (Ocrelizumab, off Label Rituximab) die einzige Option für die Therapie der primär progredienten MS. Nur Ocrelizumab hat eine Zulassung für diese Indikation (Abbildung 2). Doch auch für diese Arzneistoffe konnte eine signifikante Wirksamkeit nur für Patienten unter 45 oder 50 Jahren gezeigt werden, während mit zunehmendem Alter die Häufigkeit von Nebenwirkungen ansteigt (15, 16). Daher sollte eine Therapie mit CD20-Antikörpern nur dann bei Patienten jenseits des 50. Lebensjahres begonnen werden, wenn eine die Selbstständigkeit gefährdende Behinderung droht (1). Eine bisher gut verträgliche Therapie muss bei älteren Patienten nicht zwingend abgesetzt werden.

Abbildung 2: Therapiealgorithmus bei Patienten mit progredienter MS; modifiziert nach (1) / Foto: PZ/Stephan Spitzer
Da Arzneistoffe mit Zulassung für die sekundär progrediente MS in der Regel nur bei aktiver Erkrankung wirksam sind, muss vor Therapiebeginn eine entsprechende Klassifizierung (aktiv/nicht aktiv) erfolgen. Bei aktiver SPMS können Siponimod (oral), Beta-Interferone, Cladribin oder CD20-Antikörper zum Einsatz kommen (Abbildung 2). Am besten ist die Wirksamkeit bei jungen Patienten mit kurzer Krankheitsdauer, geringerem Behinderungsgrad, die Grundprogression überlagernden Schüben oder schneller Zunahme der Behinderung sowie hoher Entzündungsaktivität.
Bei nicht aktiver SPMS erhält der Patient keine Immuntherapie, sondern eine regelmäßige MRT-Kontrolle und klinische Verlaufskontrolle (1).
Es gibt Patienten, die vor der Immuntherapie nur eine geringe Krankheitsaktivität hatten und unter der Therapie mit Wirkstoffen der Kategorie 1 jahrelang keine Krankheitsaktivität zeigen. Falls diese es wünschen, kann nach mindestens fünf Jahren Therapiedauer ein Absetzen versucht werden. Für andere Konstellationen gibt es leider kaum Daten zur Beendigung einer Therapie, sodass die Leitlinienautoren hier keine Empfehlung geben. Arzt und Patient müssen individuell entscheiden.
Bei einem Absetzversuch sollte der Arzt nach sechs und zwölf Monaten eine Verlaufskontrolle veranlassen und bei Wiederauftreten von Krankheitsaktivität die Immuntherapie wieder verordnen.
Besonders bei Beenden der Therapie mit Fingolimod (und eventuell auch den neueren S1P-Modulatoren) und Natalizumab ist ein schnelles Wiederaufflammen der Krankheitsaktivität beschrieben. Dies müssen Arzt und Patient beim Absetzen beachten (1).

Foto: Adobe Stock/Krakenimages.com
Die neue MS-Leitlinie (1) behandelt auch die deutlich seltener auftretenden, mit MS verwandten Pathologien Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD) und MOG-IgG-assoziierte Erkrankungen.
NMOSD betreffen in Deutschland circa 1500 bis 2000 Personen, Frauen sind deutlich häufiger betroffen. Der Erkrankungsbeginn liegt später als bei MS. Meist sind das Rückenmark und der Sehnerv von den Entzündungen betroffen, die schubförmig auftreten und sich im Unterschied zur MS oft nicht vollständig zurückbilden. Kennzeichnend sind Autoantikörper gegen Aquaporin-4-Kanäle auf Astrozyten sowie hohe IL-6-Konzentrationen. Häufige Symptome sind Sehstörungen, Bewegungs- und Sensibilitätsstörungen. Spezifisch für NMOSD zugelassen sind der gegen den Komplementfaktor C5 gerichtete Antikörper Eculizumab sowie der IL-6-Rezeptor-Antikörper Satralizumab, der seit Sommer 2021 im Handel ist.
MOG-IgG-assoziierte Erkrankungen sind eine eigenständige Entität, die durch das Auftreten von Immunoglobulin-G-(IgG)-Antikörpern gegen das Myelin-Oligodendrozyten-Glykoprotein (MOG-IgG) gekennzeichnet ist. Die Symptome sind ähnlich wie bei NMOSD, allerdings sind die Manifestationen weiter gefasst und umfassen beispielsweise auch Enzephalitiden. Es gibt keine kontrollierten Studien oder spezifisch für diese Erkrankungen zugelassenen Therapeutika. Eine Behandlung mit Beta-Interferonen, Glatirameroiden oder Alemtuzumab sollte vermieden werden, da diese keine oder sogar negative Wirkungen haben.
MS-Patientinnen sollten eine Schwangerschaft möglichst in einer stabilen Phase ihrer Erkrankung planen. Die Erhaltungstherapie sollte schon bei Kinderwunsch auf Interferone oder Glatirameracetat umgestellt werden.
Abhängig von der Krankheitsaktivität muss die Frau gemeinsam mit dem Arzt entscheiden, ob die Therapie in der Schwangerschaft abgesetzt oder beibehalten werden soll. So können Beta-Interferone und Glatimeroide bei hoher Krankheitsaktivität weiterhin verabreicht werden. In schweren Fällen kann auch eine Therapie mit Natalizumab erfolgen.

Wunschkind: An MS erkrankte Frauen sollten ihre Schwangerschaft gut planen und gemeinsam mit dem Arzt die Therapie anpassen. / Foto: Adobe Stock/Ekaterina Pichukova
Dimethylfumarat sollte mit Eintritt der Schwangerschaft abgesetzt werden. Auch Teriflunomid und S1P-Modulatoren sind in der Schwangerschaft nicht geeignet. Cladribin und Mitoxantron wirken teratogen; daher müssen Männer und Frauen bis sechs Monate nach Therapieende zuverlässig verhüten. Für Alemtuzumab, Ocrelizumab und Rituximab beträgt dieser Zeitraum vier Monate nach Therapieende.
Nach dem ersten Trimenon kann ein Schub mit hoch dosierten Glucocorticoiden (Methylprednisolon oder Prednisolon) behandelt werden. Da im ersten Trimenon ein erhöhtes Risiko für Lippen-Kiefer-Gaumenspalten besteht (17, 18), sollte man Glucocorticoide nur in Ausnahmefällen geben. Bei schweren therapierefraktären Schüben oder Kontraindikationen für eine Steroidtherapie kann die Schwangere eine Immunapherese erhalten.
Stillen wirkt sich insbesondere bei Frauen, die während der Schwangerschaft keine Therapie erhielten, positiv auf die Schubrate aus. Das Apothekenteam kann die Frau daher zum Stillen ermutigen. Tritt dennoch ein Schub auf, sollte sie nach der Glucocorticoid-Gabe eine Stillpause von einer bis vier Stunden einhalten. Das Stillen muss aber nicht abgebrochen werden. Zur Verlaufstherapie können auch hier in schwereren Fällen Beta-Interferone und Glatirameracetat verwendet werden, wobei nur Erstere explizit für die Stillzeit zugelassen sind (1).
Bei etwa 5 bis 10 Prozent der Patienten manifestiert sich eine MS erst jenseits des 55. Lebensjahrs (19). In dieser Population besteht naturgemäß eine höhere Komorbidität beispielsweise in Bezug auf kardiovaskuläre Erkrankungen und andere Risikofaktoren wie ein erhöhtes Risiko für Malignome, Infektionen und psychische Komorbiditäten, die berücksichtigt werden müssen. Das allein ist jedoch kein Grund, auf eine Immuntherapie zu verzichten. Arzt, Patient und Apotheker müssen hier in besonderem Maß auf Risiken, Unverträglichkeiten und Nebenwirkungen der Medikamente achten.
3 bis 7 Prozent der MS-Erkrankungen treten bei Kindern und Jugendlichen unter 18 Jahren auf. Der Verlauf unterscheidet sich oft von dem bei Erwachsenen. Beispielsweise sind Enzephalopathien häufiger. Die Schubrate in den ersten Erkrankungsjahren ist höher, die Phase bis zum Erreichen einer klinisch relevanten Behinderung aber meist länger (1).

Die Therapie besteht aus verschiedenen Teilen. Einer davon ist die Physiotherapie. / Foto: Adobe Stock/auremar
Die bereits beschriebenen McDonald-Kriterien gelten seit 2018 auch für die Diagnostik der MS bei Kindern und Jugendlichen (20). Seit 2016 liegt eine eigene Therapieleitlinie vor, die sich derzeit überarbeitet wird (21).
Die Schubtherapie erfolgt wie bei Erwachsenen mit Glucocorticoiden, und zwar gewichtsadaptiert (20 mg/kg KG/Tag) mit einer Maximaldosis von 1000 mg/Tag. Auch die Immuntherapie ist prinzipiell ähnlich wie bei Erwachsenen, wobei nicht alle verfügbaren Arzneistoffe eingesetzt werden. Die Studienlage ist mit retrospektiven und/oder unverblindeten Studien deutlich schlechter als bei Erwachsenen. Dennoch hat die European Medicines Agency den Beta-Interferonen (zum Beispiel IFN-β1a für Kinder ab zwei Jahren) sowie Glatirameracetat (für Kinder ab zwölf Jahren) auf dieser Basis eine Zulassung erteilt (22). Diese Medikamente können bei leichten oder mittelschweren Verlaufsformen eingesetzt werden.
Aktuell laufen Kinderstudien zu Dimethylfumarat und Teriflunomid (23). Teriflunomid ist bereits für Kinder ab zehn Jahren zugelassen. Die einzige abgeschlossene, prospektive kontrollierte Studie an Kindern verglich Fingolimod mit Beta-Interferon und führte zur Zulassung von Fingolimod bei Kindern ab zehn Jahren mit hochaktiver RRMS (24).
Natalizumab, Alemtuzumab, Cladribin, Dimethylfumarat, Ocrelizumab und Rituximab sind für die Behandlung von Kindern und Jugendlichen nicht zugelassen. Für Natalizumab und Rituximab liegen jedoch retrospektive Daten vor, die eine positive Wirkung auf die Schubrate und das Auftreten neuer Läsionen nahelegen (25, 26).
Katharina Holl studierte Pharmazie in Münster und wurde dort 2013 am Institut für Pharmazeutische und Medizinische Chemie promoviert. Seitdem ist sie im Bereich der Arzneimittelzulassung in der pharmazeutischen Industrie tätig, erwarb einen Mastertitel in Drug Regulatory Affairs an der Universität Bonn und ist Fachapothekerin für Arzneimittelinformation.


