 |  | Nico Kibria |
| | Christina Lamers |
 |
12.02.2023 08:00 Uhr |

Sie schrieben Medizingeschichte: Die Forscher Frederick Banting und Charles Best isolierten das Peptidhormon Insulin aus der Bauchspeicheldrüse von Hunden. / Foto: Imago Images/Everett Collection
Die US-amerikanische Zulassungsbehörde FDA definiert Peptide als Polymere aus 40 oder weniger α-Aminosäuren, unabhängig von der Herstellung per Synthese oder durch rekombinante Expression (1).
Ein bedeutender Meilenstein der Medizingeschichte und für Peptide als Therapeutika war die Isolierung des Peptidhormons Insulin im Jahr 1921 aus der Bauchspeicheldrüse von Hunden durch Frederick Banting und Charles Best. Wenige Monate später verabreichten die beiden Forscher das Insulin erstmals einem an Typ-1-Diabetes erkrankten Jungen. Eine bis dato innerhalb von einem bis zwei Jahren tödlich verlaufende Krankheit wandelte sich durch die Insulintherapie in eine chronische Erkrankung (2).
Die ersten Peptidtherapeutika leiteten sich wie das Insulin von körpereigenen Peptidhormonen, zum Beispiel Oxytocin, ab oder wurden aus Naturstoffen isoliert wie das Ciclosporin.
Die Peptid-Festphasensynthese (SPPS) durch Merrifield 1963 war ein weiterer wichtiger Meilenstein, um die Peptidsynthese zu automatisieren und diesen Molekülen den Weg als Therapeutika zu ebnen (Abbildung 1). Jahrzehnte nach Insulin wurden die ersten synthetisch hergestellten Peptidarzneimittel zugelassen (1980 und 1978: Oxytocin und Desmopressin).
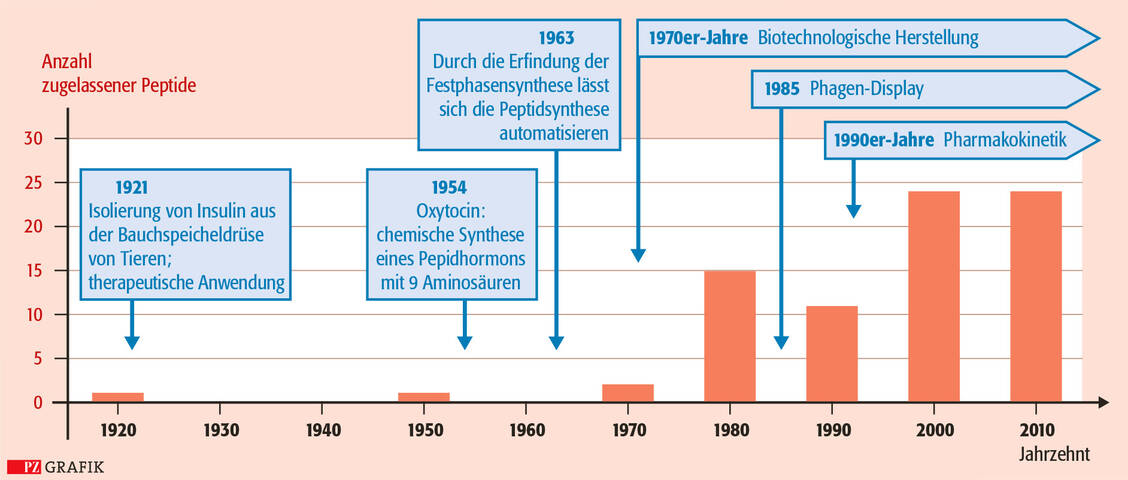
Abbildung 1: Meilensteine in der Entwicklung von Peptiden als Therapeutika. Die Festphasensynthese und das Phagen-Display haben jeweils einen Nobelpreis erhalten. Als Balkendiagramm eingetragen ist die Anzahl an zugelassenen Peptidarzneimitteln pro Jahrzehnt. / Foto: PZ/Stephan Spitzer
Erst in den 1980er-Jahren wurde durch die Fortschritte in der Biotechnologie der nächste entscheidende Schritt erreicht, der die rekombinante Herstellung von Peptiden ermöglichte. Seitdem werden diese entweder rekombinant oder synthetisch hergestellt. Jedoch brachte erst die Zulassung des HIV-Fusionsinhibitors Enfuvirtid 2003 den entscheidenden Nachweis, dass Peptide synthetisch in großem Maßstab industriell und ökonomisch sinnvoll produzierbar sind.
Seitdem und gerade in den letzten Jahren wächst der Peptidmarkt stark – dies wird sich fortsetzen. Fortschritte in der Identifizierung, Herstellung, Aufreinigung und Optimierung ebnen diesen Arzneistoffen weiter den Weg. Ihr Potenzial, das in einer hohen Bindungsaffinität und Spezifität liegt, wird in neuen Anwendungsgebieten voll ausgeschöpft.
Zurzeit sind 80 Peptide als Arzneimittel zugelassen. Mehr als 150 befinden sich in der klinischen Entwicklung, davon 60 in PhaseII oder III. Weitere 600 Peptide sind in der präklinischen Entwicklung (3).
Viele peptidische Arzneistoffe haben ihren Ursprung in natürlichen Quellen, sei es als Peptidhormon-Analoga (Insulin, Glucagon-like-peptide 1: GLP-1, Gonadorelin: GnRH, antidiuretisches Hormon: ADH) oder aus pflanzlichen und tierischen Quellen. So stellen Abwehrstrategien von Mikroorganismen, Pflanzen und Tieren gegen Pathogene oder Fressfeinde eine reiche Quelle von potenten bioaktiven Peptiden dar.
Weitere Beispiele: Das Gift der Kegelschnecke diente als Grundlage für die Entwicklung des zyklischen Peptids Ziconotid. Dieses wird bei chronischen Schmerzen intrathekal angewandt und ist 1000-fach stärker wirksam als Morphin, ohne zu Abhängigkeit oder Sucht zu führen (4). Exenatid wurde aus dem Gliamonster isoliert und wird wegen der zuckersenkenden Wirkung als Diabetesmedikament eingesetzt.
Eine große Klasse stellen antimikrobielle Peptide wie die Glyko- und Lipopeptide dar. Wichtige Vertreter sind Daptomycin, Dalbavancin, Ramoplanin, Vancomycin, Oritavancin, Telavancin, Caspofungin, Micafungin und Anidulafungin. All diesen antimikrobiell wirksamen Peptiden ist gemeinsam, dass sie nicht ribosomaler Natur sind, das heißt: Sie sind zyklisch und mit untypischen Strukturelementen ausgestattet. Dies führt zu einer hohen Stabilität gegen Peptidasen und einer außergewöhnlichen Potenz. Ein weiteres wichtiges Beispiel eines Arzneistoffs aus natürlicher Quelle ist das Immunsuppressivum Ciclosporin.
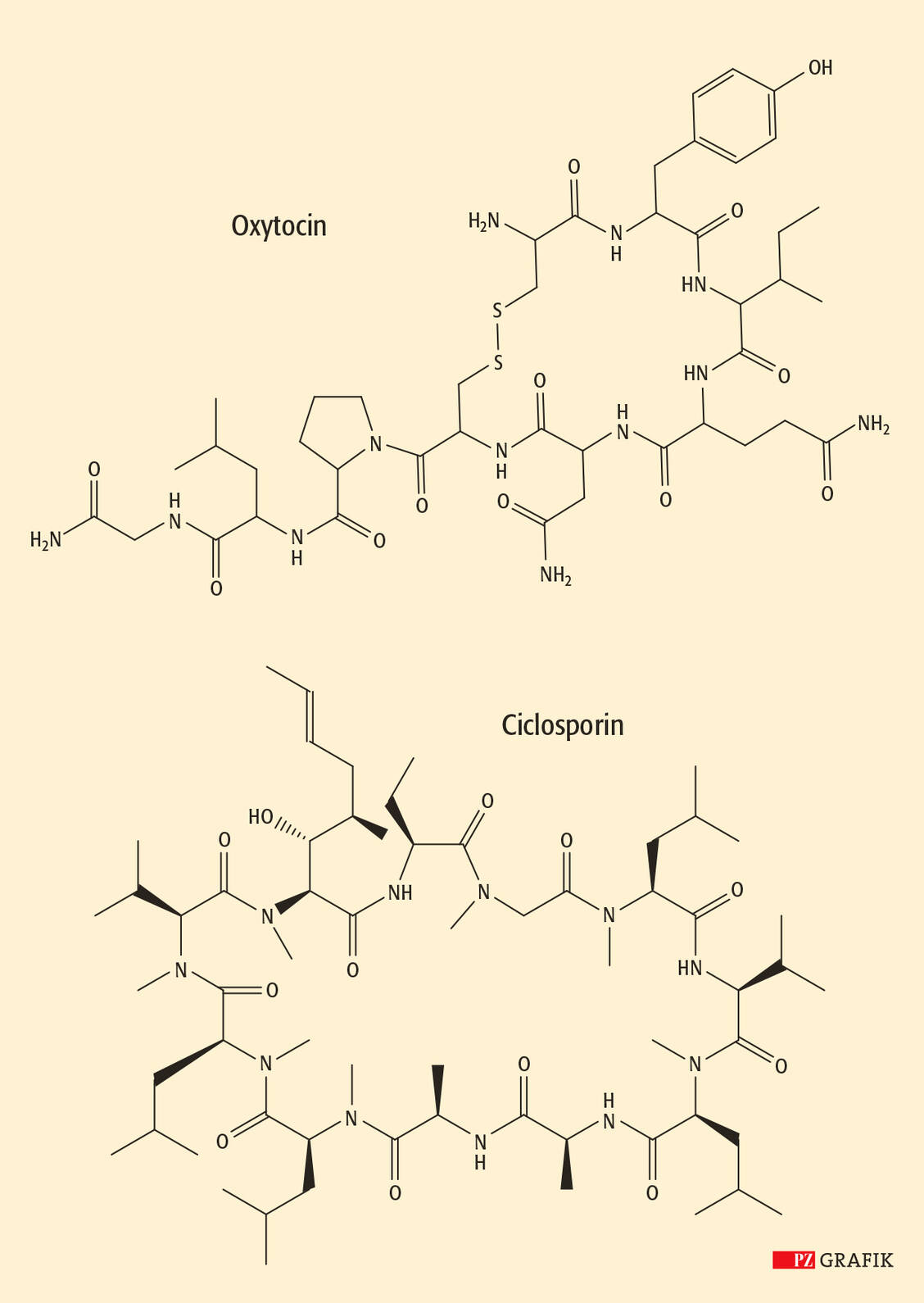
Abbildung 2: Oxytocin und Ciclosporin als Beispiele etablierter zugelassener Peptidarzneistoffe aus natürlichen Quellen / Foto: PZ
Peptide stellen eine besondere Arzneistoffklasse mit hoher Wirkpotenz und Selektivität dar. Jedoch haben sie gewisse Grenzen, die besonders ihre pharmakokinetischen Eigenschaften betreffen (Tabelle 1).
Limitierend für die breite Anwendung als Arzneimittel ist die fehlende orale Bioverfügbarkeit der meisten Peptide. Bei oraler Applikation ist ihre chemische (Magensäure) und enzymatische Stabilität herausgefordert. Im Magen und Dünndarm zerlegen Peptidasen (Magen: Pepsin; Dünndarm: Trypsin und Chymotrypsin) Proteine und Peptide durch enzymatische Spaltung der Peptidbindung.
Falls ein Peptid diese Hürden durch ausreichende Stabilität oder eine entsprechende Formulierung überwindet, verhindert die mangelnde Membranpassage in den Blutkreislauf die Wirksamkeit. Aufgrund der mangelnden Membranpassage wird bisher der Großteil der Peptidtherapeutika parenteral verabreicht.
| Einschätzung | Beispiele |
|---|---|
| Vorteile, Chancen | hohe Affinität und Potenz, daher geringe Dosis nötighohe Selektivität, daher kaum Off-target-Effektegeringe Toxizität und in der Regel keine toxischen Metabolitegeringe Immunogenität im Vergleich zu Antikörpern und Proteinenkeine Akkumulation im Gewebe |
| Nachteile, Grenzen | geringe chemische und proteolytische Stabilität im Gastrointestinaltraktin der Regel nicht membrangängigin der Regel orale BioverfügbarkeitLöslichkeitsprobleme geringe Plasmastabilitäthohe glomeruläre FiltrationProduktion zum Teil kostenintensiv |
Zur Optimierung der Eigenschaften von Peptiden haben sich mehrere Strategien etabliert, die auf einer direkten chemischen Modifikation (5, 6) oder einer Formulierung basieren. Durch chemische Veränderung der Peptidstruktur soll der Abbau durch Peptidasen verlangsamt oder verhindert werden. Hier ist die Zyklisierung der Peptide weit verbreitet, die zu sterisch anspruchsvollen Strukturen führt, die von den Peptidasen nicht mehr erkannt werden.
Chemisch eignen sich der C- und N-Terminus ebenso wie die Seitenketten zur Zyklisierung. Bei einer Verknüpfung von C- und N-Terminus spricht man von einer »Head-to-Tail«-Zyklisierung. Viele Peptidasen arbeiten als Exopeptidase und brauchen daher für den Abbau einen freien N- oder C-Terminus, der durch die Head-to-Tail-Zyklisierung wegfällt.

Wie Insulin müssen die meisten peptidischen Arzneistoffe, so auch GLP-1-Analoga, gespritzt werden. / Foto: Adobe Stock/RFBSIP
Weiterhin können funktionelle Gruppen der Seitenketten mit dem N- oder C-Terminus reagieren (»Head-to-Sidechain«, »Sidechain-to-Tail«). Auch Seitenketten sind miteinander zyklisierbar; typisch ist hier die Bildung einer Disulfid-Brücke zwischen zwei Cysteinen (7).
Verwendet man Spiegelbildisomere (Enantiomere) der L-Aminosäuren zur Synthese, resultieren Peptide mit gleichen physikochemischen Eigenschaften, die aber nicht von Peptidasen erkannt werden. Auch eine N-Methylierung unterbindet durch sterische Hinderung die enzymatische Hydrolyse der Peptidbindung.
Eine weitere Limitation von Peptidtherapeutika ist deren kurze Halbwertszeit in vivo durch eine ausgeprägte renale Filtration (Tabelle 1). Diese kann reduziert werden durch die Konjugation des Peptids mit Makromolekülen wie PEG-Polymeren. Eine weitere häufig genutzte Strategie ist die Konjugation mit Fettsäuren, die eine Bindung an Albumin vermitteln und somit das Peptid vor der renalen Filtration schützen (17). Beispiele sind die GLP-1-Analoga Liraglutid (C16-Fettsäure) und Semaglutid (PEG-Spacer und C18-Dicarbonsäure) sowie Insulin Detemir (C14-Fettsäure) und Insulin Degludec (C16-Fettsäure). Bei all diesen Peptiden führt die Modifikation zu einer längeren Zirkulationshalbwertszeit. Sie müssen daher nur einmal täglich bis zu einmal wöchentlich gespritzt werden.
Anstatt das Peptid chemisch zu verändern, was zu pharmakodynamischen Einbußen führen kann, ist eine direkte Hemmung der Peptidasen durch Inhibitoren möglich (5). Inhibitoren wie Aprotinin (Trypsin-Hemmer), Amastatin und Bestatin (Hemmer der Aminopeptidase) können mit dem Peptidwirkstoff coformuliert werden. Diese Strategie zeigt in vitro gute Ergebnisse, doch die Effektivität in vivo ist limitiert.
Der Zusatz von Penetrationsverstärkern soll die Resorption im Darm erhöhen. Weitere arzneiformbezogene Konzepte werden beispielsweise mit Mikropartikeln oder Nanopartikeln realisiert (5–12). Jedoch gibt es noch keine generell einsetzbare Lösung für die mangelnde Bioverfügbarkeit von Peptiden.
Der oral verfügbare GLP-1-Agonist Semaglutid hat 2020 die Zulassung erhalten. Hierbei ist das Peptid mit dem Fettsäurederivat SNAC (Sodium-N- (8-[2-hydroxybenzol]amino)caprylat) in einer Tablette formuliert. SNAC sorgt im Magen für einen lokalen pH-Anstieg; dieser schützt vor dem proteolytischen Abbau durch nicht optimale pH-Bedingungen für Pepsin. Gleichzeitig verbessert SNAC die Löslichkeit des Wirkstoffs. Die Aufnahme erfolgt im Magen, wahrscheinlich über die transzelluläre Route. Die orale Bioverfügbarkeit beträgt 1 Prozent, wobei starke Schwankungen zwischen Patienten und in Abhängigkeit von der Nahrungsaufnahme auftreten. Daher sollte der Patient die Tablette morgens auf leeren Magen 30 Minuten vor einer Mahlzeit einnehmen.
Interessant ist, dass mit der peroralen Dosis von 14 mg/Tag eine Plasmakonzentration erreicht wird wie durch die einmal wöchentliche subkutane Gabe von 0,5 mg Semaglutid (13, 14).
Ein bemerkenswertes Peptid ist Ciclosporin, das als zyklisches Peptid aus dem Schlauchpilz Tolypocladium inflatum isoliert wurde (Abbildung 2). Dieser Wirkstoff wird seit Jahrzehnten als Immunsuppressivum angewendet. Die orale Bioverfügbarkeit von Ciclosporin liegt im Mittel bei 34 Prozent und wird durch N-Methylierung, D-Aminosäuren sowie die zyklische Struktur erreicht.
Auch die Formulierung ist ein essenzieller Faktor. Die anfänglich verwendete Lösung in Oliven- oder Kornöl wurde durch ein Self-Emulsifying-Drug-Delivery-System (SEDD) ersetzt. Da auch diese Emulsion keine gut reproduzierbare Bioverfügbarkeit liefert, wurde sie später durch das Self-Micro-Emulsifying-Drug-Delivery-System (SMEDD) abgelöst. Dieses System ist eine einphasige Lösung mit hohem Emulgatoranteil und verschiedenen Co-Solventien. Im Darm wird das System freigesetzt und bildet im wässrigen Darmlumen eine Mikroemulsion mit hervorragenden Lösungseigenschaften und einer reproduzierbaren Bioverfügbarkeit. Das Produkt wird als Weichgelatinekapsel vertrieben (15, 16).
Viele Jahrzehnte war die Entwicklung von Peptidarzneistoffen geprägt von der Identifizierung und Isolierung natürlicher Peptide. Im Vergleich zu niedermolekularen Wirkstoffen etablierte sich die rationale Entwicklung von Peptidarzneistoffen aufgrund ihrer hohen strukturellen Komplexität langsam.
Die Fortschritte in der Strukturaufklärung der letzten Jahrzehnte ermöglichen jetzt die rationale Entwicklung von Peptiden, die sich von Proteinsekundärstrukturen ableiten. Häufig handelt es sich um Inhibitoren sogenannter Protein-Protein-Interaktionen, beispielsweise die Tumorsuppressor-Interaktion von MDMX/p53 durch ALRN-6924 (in PhaseII). Hierbei werden Strukturelemente eines Proteins, zum Beispiel α-Helices, die maßgeblich an einer Protein-Interaktion zur Bildung des funktionellen Multiproteinkomplexes beteiligt sind, mittels synthetischer Peptide nachgeformt. Somit wird die Interaktion der beiden Proteine kompetitiv durch das Peptid gehemmt, was im Fall von MDMX/p53 zur Freisetzung des Tumorsuppressors p53 und somit zur Einleitung der Apoptose führt.
Ein gänzlich neuer Ansatz sind Technologien, die aus einer großen Substanzbibliothek potenzielle Wirkstoffkandidaten herausfiltern. Hervorzuheben sind Methoden, die eine genetische Codierung der Peptidsequenz nutzen und somit die Selektion einer Hit-Substanz aus Millionen Substanzen ermöglichen.
Das Phagen-Display wurde 1982 von George P. Smith und Greg Winter entwickelt und ursprünglich zur Identifizierung und Entwicklung von Antikörpern genutzt (18). Wie funktioniert diese Methode?
Bakteriophagen wie die abgebildeten M13-Phagen sind wichtige Helfer bei der Suche nach neuen Peptidarzneistoffen. / Foto: Shutterstock/Love Employee
Zunächst werden in die DNA von Phagen (Bakterienviren) Basensequenzen eingefügt, die für eine große Vielfalt von potenziell interessanten Antikörpern oder Peptiden codieren. Diese Sequenzen werden so eingefügt, dass die Peptide mit einem Hüllprotein des Phagen verknüpft sind und somit auf dessen Oberfläche präsentiert werden. An jeder Position der Sequenz kann jede der proteinogenen 20 Aminosäuren eingefügt werden, sodass jede mögliche Kombination in der Bibliothek enthalten ist. Nach Infektion von Bakterien (E. coli) durch die modifizierten Phagen werden diese in E. coli vervielfältigt und im Anschluss aufgereinigt.
Die Phagen-Protein-Bibliothek wird nun mit einem Zielprotein, das immobilisiert vorliegt, zum Beispiel einem Rezeptor, inkubiert. Aus der Vielzahl der exprimierten diversen Peptide können einige an das immobilisierte Target andocken – das sind die gesuchten Sequenzen; nicht-bindende Phagen werden weggewaschen. Durch eine erneute Vervielfältigung der bindenden Phagen können interessante Peptidkandidaten angereichert und anschließend kann durch Sequenzierung der Phagen-DNA die Sequenz des Peptids identifiziert werden.
Eine ähnliche In-vitro-Selektionsmethode verwendet mRNA als genetischen Informationsträger der Peptidbibliothek. Bei dieser Methode, dem mRNA-Display, wird die mRNA während der Translation kovalent an das gerade gebildete Peptid gebunden. Die Selektion erfolgt wieder gegen ein immobilisiertes Protein und die Sequenzierung der mRNA ermöglicht die Identifizierung des gesuchten Peptids (19). Da hierbei kein lebendes System (wie E.coli), sondern nur aufgereinigte Komponenten verwendet werden, können auch Peptide mit nicht-proteinogenen Aminosäuren in der Bibliothek enthalten sein. Dies kann aufgrund von deren besseren pharmakokinetischen Eigenschaften und der Möglichkeiten zur Zyklisierung vor der Selektion vorteilhaft sein.
Doch auch mit Phagen-Display können zyklische Peptide untersucht und gefunden werden: entweder durch die Zyklisierung über Disulfid-Brücken oder die Verwendung von chemisch modifiziertem Phagen-Display, bei dem Peptide durch chemische Reaktionen auf den Phagen zyklisiert werden (20).
Erste zyklische Peptidtherapeutika, die mit diesen Methoden entwickelt wurden, sind in klinischer Entwicklung oder bereits zugelassen. Pegcetacoplan, ein Disulfid-zyklisiertes Peptid, wurde durch Phagen-Display identifiziert und nachfolgend auf Affinität und Stabilität optimiert. Es fungiert als Protein-Protein-Inhibitor und verhindert die Aktivierung des Komplementproteins C3, das im Blut zirkuliert. Zugelassen ist es seit Mai 2021 zur Therapie der paroxysmalen nächtlichen Hämoglobinurie (Aspaveli®). Durch Konjugation mit einem PEG-Makropolymer wird die Zirkulationshalbwertszeit durch verzögerte renale Filtration erhöht (21).
Auch Zilucoplan ist ein Inhibitor des Komplementsystems auf der Stufe des C5-Proteins und verhindert damit den Angriff des Körpers auf neuromuskuläre Verbindungen. Dieses zyklische Peptid wurde durch mRNA-Display entwickelt und befindet sich in Phase II und III der klinischen Entwicklung zur Therapie von Myasthenia gravis und amyotropher Lateralsklerose (ALS). Auch Zilucoplan ist konjugiert mit PEG-Einheiten und einer Fettsäure, um die Zirkulation zu verlängern (22). Es wird subkutan appliziert.
Derzeit befinden sich knapp 60Peptide, die sich in unterschiedliche Gruppen einteilen lassen, in der späten klinischen Phase (Tabelle 2). Die größte Gruppe besteht aus Peptiden, die mittels klassischer Target-Interaktion einen pharmakologischen Effekt erzeugen. Ein Großteil ist – wie GLP-1-Analoga – den Peptidhormon-Analoga zuzuordnen, doch es zeichnet sich bezüglich der Indikationen ein neuer Trend ab.
Seit 2018 sind Peptid-Radiotherapeutika (Theranostika) zugelassen. Weitere Kandidaten werden zur Diagnostik und Therapie von soliden Tumoren entwickelt. Mit einem ähnlichen Prinzip arbeiten auch Peptid-Drug-Konjugate, die einen zytotoxischen Wirkstoff durch die Affinität der Peptidkomponente zielgerichtet zum Tumor bringen können.
Die dritte Gruppe in klinischer Entwicklung besteht aus Peptidvakzinen, die zur Therapie von Krebserkrankungen entwickelt werden (Kasten).
| Substanzname | Indikation | Klinische Phase |
|---|---|---|
| Peptidarzneistoff | ||
| Zilucoplan (RA101495) | Myasthenia gravis, amyotrophe Lateralsklerose, paroxysmale nächtliche Hämoglobinurie | II/III |
| QL0911 | Chemotherapie-induzierte Thrombozytopenie | III |
| Apraglutid | Kurzdarmsyndrom | III |
| Glepaglutid (ZP1848) | Kurzdarmsyndrom | III |
| Solnatid | akutes Lungenversagen | II |
| Apelin + Relaxin | Typ-2-Diabetes mit kardiovaskulärem Risiko | II |
| Radiotherapeutikum | ||
| 177Lu-DOTA-Tyr3-Octreotat | neuroendokrine Tumoren im Pankreas | II |
| 68Ga-FAP-2286 | solide Tumoren | II |
| therapeutische Vakzine | ||
| GLSI-100 | bei HER2-positivem Brustkrebs | III |
| IO102-IO103 | bei nicht kleinzelligem Bronchialkarzinom, Melanom | Phase II/III |
Foto: Adobe Stock/PRB ARTS
Krebszellen tragen durch Mutation oder erhöhte Expression sogenannte Peptidepitope auf ihrer Oberfläche, die sich von gesunden Zellen unterscheiden. Diese Epitope können identifiziert, synthetisch hergestellt und als Vakzine formuliert werden, sodass sie von dem Haupthistokompatibilitätskomplex II (MHC II) durch antigenpräsentierende Zellen (dendritische Zellen) präsentiert werden und somit CD4+-T-Zellen aktivieren. Dies führt zu einer Th1-Antwort mit INF-γ-Freisetzung, Aktivierung von Makrophagen und Unterstützung der zytotoxischen T-Zell-Funktion. Ein Teil der Peptidepitope kann auch über MHC I den CD8+-T-Zellen präsentiert werden, was zu einer direkten zellvermittelten Zytotoxizität gegenüber Peptidepitop-tragenden Zellen führt.
Vielversprechende Peptidvakzine werden vor allem in der Therapie von Brustkrebs, Gliomen und Melanomen getestet und befinden sich aktuell in Phase I und II (26–28).
Bisher gibt es weder von der FDA, der EMA oder dem International Council for Harmonisation (ICH) festgesetzte Richtlinien, die spezifisch für Peptide im Zulassungsverfahren gelten. So wird bisher bei jedem Peptidwirkstoff individuell festgesetzt, welche Analytik und Grenzwerte bezüglich des Nachweises von Verunreinigungen erforderlich und akzeptabel sind. Um diese regulatorische Lücke zu schließen, veröffentlichte die EMA ein Konzeptpapier, das die Erstellung von Richtlinien für die Entwicklung und Herstellung synthetischer Peptide einfordert (23).
Besonders die Festphasensynthese der Peptide unterscheidet sich deutlich von der Synthese niedermolekularer Verbindungen. So ist zum Beispiel eine chromatografische Aufreinigung notwendig, die besondere Anforderungen stellt. In der Synthese können Peptidsequenzen entstehen, denen eine oder einige Aminosäuren im Vergleich zum Hauptprodukt (Deletion) fehlen; diese Nebenprodukte können sehr ähnliche physikochemische Eigenschaften wie das Hauptprodukt haben. Daher empfiehlt das Konzeptpapier die Verwendung einer zweiten orthogonalen Reinigungsmethode, um die Verunreinigungen ausreichend vom aktiven Inhaltsstoff zu trennen.

Die meisten Peptidarzneistoffe, zum Beispiel antimikrobiell wirksame Peptide, werden parenteral appliziert. / Foto: Adobe Stock/Gorodenkoff
Natürlich muss auch die Identität nachgewiesen werden. Dafür sind folgende Methoden vorgesehen: HPLC-Analytik mit der Co-Injektion eines Standards, der nur in einem Peak resultieren darf, sowie Massenspektrometrie (MS), wobei hier Stereoisomere nicht identifiziert werden können. Auch die Aminosäuren-Analyse ist gefordert, die nach Derivatisierung der einzelnen Aminosäuren mittels chiraler Gaschromatografie erfolgt. Bei Peptiden unter zehn Aminosäuren kann die NMR-Analytik sinnvoll sein; bei längeren Peptiden empfiehlt sich das Peptid-Mapping (enzymatischer Verdau und Analyse der Peptidfragmente mittels MS).
Basierend auf dem Herstellungsprozess (Synthese oder rekombinante Expression) werden unterschiedliche Analysen auf Verunreinigungen gefordert. Da größere Peptide Sekundärstrukturen ausbilden können, die relevant für die Wirksamkeit sind, muss die Sekundärstruktur nachgewiesen werden. Dazu eignen sich Circulardichroismus, Fourier-Transform-Infrarotspektrometrie und Kernspinresonanz-Spektroskopie (NMR) (24, 25).
Ein wichtiger Sicherheitsfaktor bei Peptiden ist die Bildung von Aggregaten, die ab einer bestimmten Größe eine Immunantwort und Antikörperproduktion auslösen können. Aggregate können durch Temperatur, Licht, Schütteln, Oberflächenkontakt und pH-Anpassungen während Herstellung, Verarbeitung, Transport oder Lagerung entstehen. Die Zusammenlagerung kann durch Peptidmodifikationen initiiert werden, die durch Oxidation, Deamidierung und Änderungen in der Primärsequenz auftreten können, sowie durch Hilfsstoffe und Primärpackmittel. Zur Identifizierung der Aggregate eignen sich orthogonale analytische Methoden wie Größenausschluss-Chromatografie, Ultrazentrifugation und dynamische Lichtstreuung.
Aktuell werden die meisten Peptidarzneimittel parenteral in speziellen Applikationssystemen (Pens) oder Infusionslösungen (aus einem Konzentrat verdünnt) verabreicht. Dies stellt besondere Anforderungen an den Herstellungsprozess (Sterilität) sowie die Lagerung und Anwendung durch den Patienten. Dabei sind Peptide im Allgemeinen stabiler als Proteinarzneistoffe, müssen aber dennoch über längere Zeit kühl gelagert werden. Wichtig ist, dass die Patienten die Lagerungs- und Anwendungshinweise kennen (Beispiele in Tabelle 3).
| Peptidwirkstoffe (Beispiele) | Darreichungsform | Hinweise zur Lagerung und Anwendung |
|---|---|---|
| Insulin-Analoga: Glargin, Detemir, Lispro, Glulisin, Aspartat | Fertigpen (Lösung zur subkutanen Anwendung) | lichtgeschützt lagernvor Anbruch im Kühlschrank lagern (2 bis 8 °C), nach Anbruch bei Raumtemperatur (<25 °C)nicht einfrierennicht mit aufgesetzter Nadel lagernnicht stark schütteln oder aufschlagenPen mit feuchtem Tuch reinigen, nicht durchnässen oder waschen |
| GLP-1-Analoga: Exenatid, Liraglutid, Semaglutid, Dulaglutid | Fertigpen (Suspension zur subkutanen Anwendung) | lichtgeschützt und im Kühlschrank lagern, außerhalb maximal 14 Tage (<30 °C)nicht einfrierenmanche Pens müssen flach gelagert werdenindividuelle Haltbarkeit nach Anbruch (2 bis maximal 6 Wochen) |
| Ciclosporin | Weichgelatinekapseln | kühl und trocken lagern (maximal 25 °C)beim Öffnen des Blisters ist ein charakteristischer Geruch wahrzunehmen, der jedoch unbedenklich ist |
| Ciclosporin | Augentropfen (Emulsion) | nicht einfrierenkühl und trocken lagern (maximal 25 °C)vor Licht schützen |
| Ciclosporin | Emulsion zur oralen Einnahme | bei 15 bis 30 °C lagern, nicht im Kühlschrankkein Problem, falls Flocken und kleine Ablagerungen entstehen: es handelt sich um Hilfsstoffe, nicht um den Wirkstoff |
| Ciclosporin | Infusionslösung (Konzentrat) | nicht unverdünnt anwendenzur Herstellung der Infusionslösung Glasampullen verwendensobald Ampulle geöffnet ist, unverzüglich im Kühlschrank lagernnach Zubereitung innerhalb von maximal 24 h verwenden |
Peptide galten trotz ihrer hohen Wirkpotenz und Selektivität lange Zeit nicht als ideale Therapeutika. Erst technische Fortschritte in der Festphasensynthese, die biotechnologische Produktion und die Etablierung von Strategien zur Verbesserung der pharmakokinetischen Eigenschaften ab den 1980er/90er-Jahren haben das Potenzial nutzbar gemacht. Seitdem steigt die Zahl an neu zugelassenen Peptidarzneistoffen deutlich an. Neues Potenzial bieten chemisch modifizierte zyklische Peptide. Neue Anwendungsgebiete eröffnen sich mit den Peptidvakzinen und der Theranostik, die besonders die Krebstherapie revolutionieren können.
Eine weiterhin unbezwingbare Hürde für die meisten Peptide ist die Plasmamembran, sodass bisher selten intrazelluläre Targets adressiert werden konnten. Doch auch hier sind Strategien in der Entwicklung wie zellpenetrierende Peptide, die in die Zelle aufgenommen werden. Aufgrund dieser Entwicklungen werden in Zukunft vermehrt Peptide als Arzneimittel zugelassen werden.
Nico Kibria studierte Pharmazie an der Universität Leipzig und absolvierte sein zweites Staatsexamen im September 2022. Aktuell fertigt er im Rahmen seines Praktischen Jahres eine Diplomarbeit im Arbeitskreis von Juniorprofessorin Christina Lamers an. Seine Diplomarbeit widmet sich potenziellen Peptid-Wirkstoffkandidaten als Inhibitoren des Komplementsystems des Menschen.
Christina Lamers studierte Pharmazie an der Goethe-Universität Frankfurt am Main und erhielt 2009 die Approbation als Apothekerin. Es folgte die Promotion am dortigen Institut für Pharmazeutische Chemie. Von 2015 bis 2017 forschte Lamers als Marie-Curie-Fellow-Postdoc an der Ecole Polytechnique Federal Lausanne zum Phagen-Display und zu Peptidinhibitoren der intrinsischen Koagulation. Anschließend war sie als Postdoc und Lehrbeauftragte an der Universität zu Basel mit Forschungsschwerpunkt Identifizierung und Optimierung zyklischer Peptidtherapeutika tätig. Seit Mai 2022 ist Dr. Lamers Juniorprofessorin für Pharmazeutische und Medizinische Chemie an der Universität Leipzig.


