 |  | Robert Fürst |
| | Ilse Zündorf |
 |
30.07.2020 10:00 Uhr |

Die Gerinnungskaskade ähnelt einem Dominospiel: Jeder Gerinnungsfaktor agiert als Protease und aktiviert dadurch den nachgeschalteten Faktor. / Foto: iStock/BrianAJackson
Die berühmteste von Hämophilie betroffene Familie waren vermutlich die Nachkommen von Queen Victoria. Bekannt war die Erkrankung allerdings schon sehr viel früher. Bereits im 2. Jahrhundert verfügte der Rabbi Judah, dass ein jüdischer Junge nicht beschnitten werden sollte, falls bereits zwei seiner Brüder vorher bei der Zeremonie verblutet waren.
Schon früh wusste man, dass die erhöhte Blutungsneigung rezessiv vererbt wird und vor allem männliche Nachkommen betrifft. Der zugrundeliegende Pathomechanismus konnte jedoch erst identifiziert werden, als Mitte des 20. Jahrhunderts die Biochemie der Blutgerinnung aufgeklärt war. Die Gerinnungsfaktoren erhielten römische Zahlen (Tabelle 1), wobei die Nummerierung nicht der Abfolge in der Gerinnungskaskade entspricht.
| Kürzel | Beschreibung | Fallprävalenz* in der allgemeinen Bevölkerung |
|---|---|---|
| FI | Fibrinogen | 1 in 1 Million |
| FII | Prothrombin | 1 in 2 Millionen |
| FIII | Prothrombinase, Gewebsthrombokinase, Tissue-(Gewebe-)Faktor (TF) | |
| FIV | Ca2+ | |
| FV | Proaccelerin, Plasma-Ac-Globulin | 1 in 1 Million |
| FVI | Accelerin | |
| FVII | Proconvertin, Prothrombinogen, Co-Thromboplastin | 1 in 500.000 |
| FVIII | antihämophiles Globulin A (AHG A), antihämophiler Faktor A (AHFA) | 6 in 100.000 Jungen |
| FIX | antihämophiles Globulin B (AHG B), antihämophiler Faktor B (AHF B) | 1 in 100.000 Jungen |
| FX | Stuart-Prower-Faktor, Thrombokinase | 1 in 1 Million |
| FXI | Rosenthal-Faktor, antihämophiles Globulin C | 1 in 1 Million |
| FXII | Hagemann-Faktor | |
| FXIII | Fibrin-stabilisierender Faktor (FSF) | 1 in 3 Millionen |
| *Prävalenzen beziehen sich auf die schweren Formen der Krankheiten, die durch homozygote oder zusammengesetzt heterozygote Genmutationen hervorgerufen werden. |
Verletzen wir uns und bluten, kommt es durch Aktivierung der Thrombozyten zunächst zu einer primären oder auch zellulären Hämostase, also der Bildung eines Thrombozytenpfropfs. Dieser ist in Form des sogenannten weißen Thrombus allerdings nicht allzu stabil. Erst wenn auch die sekundäre oder plasmatische Hämostase einsetzt, kommt es zur effizienten Blutstillung. Diese plasmatische Hämostase ist auch als Gerinnungskaskade bekannt und startet durch verletztes Endothel, dessen Zellen auf der Oberfläche den Gewebefaktor (Tissue Factor, TF) tragen. In Anwesenheit von Calciumionen wird eine Aktivierungsabfolge in Gang gesetzt, die zunächst mit dem Faktor VII beginnt. In den nachfolgenden Schritten kommen nach und nach die anderen Gerinnungsfaktoren zum Einsatz, wobei meist aus einer inaktiven Vorstufe durch Abspaltung eines kurzen Peptids eine aktive Proteinvariante gebildet wird. Ähnlich einem Dominospiel agiert das jeweilige Protein selbst wieder als Protease, schneidet den nachgeschalteten Gerinnungsfaktor und aktiviert ihn dadurch.
Während der Initiationsphase der Gerinnung entstehen nach Aktivierung der Faktoren VII und X zunächst geringe Mengen an aktivem Thrombin, das wiederum Fibrinogen schneidet und damit für die Bildung des Fibringerinnsels benötigt wird. Der Prozess wird dramatisch beschleunigt und es wird wesentlich mehr Thrombin bereitgestellt, sobald die als Kofaktoren wirkenden Gerinnungsfaktoren VIII und V ebenfalls aktiviert werden. Aktivierter Faktor VIII (FVIIIa) bildet zusammen mit FIXa einen Komplex und erhöht die Enzymaktivität von FIXa, was zu wesentlich mehr FXa führt. Zusammen mit FVa aktiviert FXa anschließend Prothrombin zu Thrombin (Abbildung). Infolgedessen wird ausreichend viel Fibrinogen gespalten, sodass ein stabiler Thrombus entsteht und die Blutung gestoppt wird. Normalerweise ist der Prozess innerhalb weniger Minuten abgeschlossen.
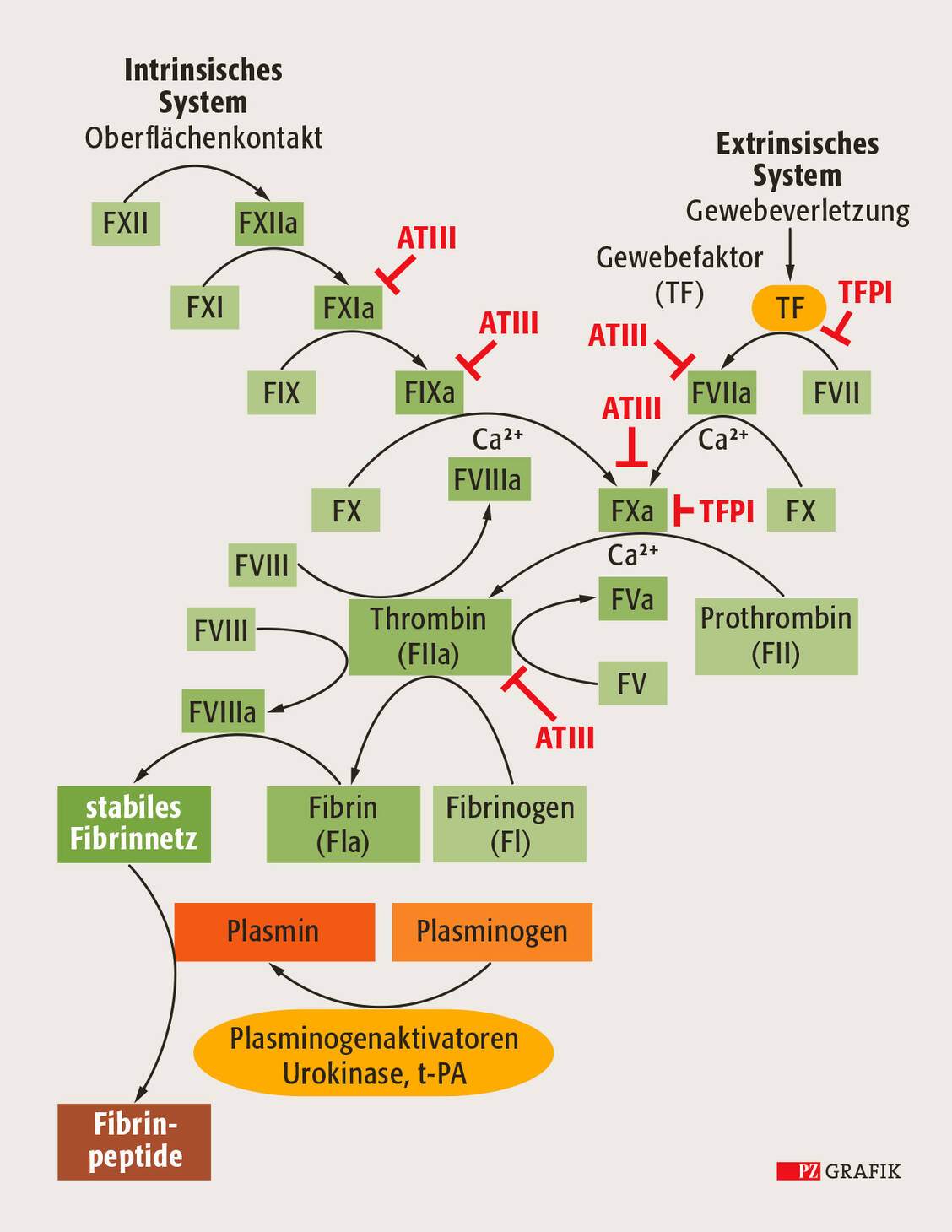
Abbildung: Schematische Darstellung der Gerinnungskaskade. Die Blutgerinnung kann auf zwei unterschiedliche Wege induziert werden: Intravasale plasmatische Faktoren aktivieren den intrinsischen Weg, während extravasale Gewebefaktoren den extrinsischen Weg initiieren. Das Prinzip der Gerinnungskaskade beruht auf der Aktivierung inaktiver Vorstufen durch Abspaltung kleiner Peptidreste aus der inaktiven Vorstufe. Gekennzeichnet werden die aktiven Varianten der Gerinnungsfaktoren durch das Suffix »a«. Sie wirken als aktive und spezifische Proteasen auf den jeweils nachfolgenden, inaktiven Gerinnungsfaktor. Als Beispiele für spezifische Inhibitoren der Gerinnungskaskade sind Antithrombin III (ATIII) und der Gewebefaktorinhibitor (Tissue factor pathway inhibitor, TFPI) mit ihren Interaktionspartnern gezeigt. / Foto: PZ
Bei wenigen Menschen ist jedoch eine erhöhte Blutungsneigung zu beobachten, die auf einen Mangel einer der beteiligten Gerinnungsfaktoren zurückzuführen ist. Derartige Koagulopathien gehören zu den seltenen Erkrankungen, das heißt, dass es in Europa weniger als 5 Fälle pro 10.000 Einwohner gibt. Die häufigsten Krankheitsbilder sind Hämophilie A und B, die durch den Mangel an FVIII beziehungsweise FIX ausgelöst werden (Tabelle 2). In Deutschland gibt es circa 4000 Patienten mit Hämophilie A und ungefähr 700 mit Hämophilie B.
| Schwere der Blutung/Art des chirurgischen Eingriffs | Benötigter Plasmaspiegel [%] | Häufigkeit der Dosierung [Stunden]/Behandlungsdauer [Tage] |
|---|---|---|
| Blutungen: | ||
| Gelenkblutungen im Frühstadium, Muskelblutungen, Blutungen im Mundbereich | 20 – 40 | Injektion alle 12 bis 24 Stunden wiederholen. Mindestens 1 Tag, bis die (durch Schmerzen erkennbare) Blutung sistiert bzw. Wundheilung erreicht ist. |
| Ausgeprägtere Gelenkblutungen, Muskelblutungen oder Hämatome | 30 – 60 | Injektion alle 12 bis 24 Stunden für 3 bis 4 Tage oder länger wiederholen, bis die Schmerzen und akute Behinderungen beseitigt sind. |
| Lebensbedrohliche Blutungen | 60 – 100 | Injektion alle 8 bis 24 Stunden wiederholen, bis die Gefahr vorüber ist. |
| Chirurgische Eingriffe | ||
| Kleinere Eingriffe einschließlich Zahnextraktionen | 30 – 60 | Einzelinfusion plus orale antifibri-nolytische Therapie innerhalb 1 Stunde ist bei ca. 70 % der Fälle ausreichend. Injektion alle 24 Stunden. Mindestens 1 Tag, bis die Wundheilung erreicht ist. |
| Größere Eingriffe | 80 – 100 (prä- und postoperativ) | Injektion alle 8 bis 24 Stunden wiederholen, bis ausreichende Wundheilung erreicht ist. Dann für mindestens weitere 7 Tage einen Faktor VIII-Spiegel von 30 % bis 60 % aufrechterhalten. |
Klinisch-symptomatisch lassen sich die beiden Hämophilie-Formen nicht unterscheiden. Beide Proteine sind auf dem X-Chromosom kodiert, weshalb Frauen extrem selten davon betroffen, aber Überträger der beiden Erbkrankheiten sind. Der Schweregrad einer Hämophilie wird nach der erhaltenen Restaktivität des Kofaktors beziehungsweise Enzyms in schwere (<1 I.E./dl), mittelschwere (1 bis 5 I.E./dl) und leichte (5 bis 40 I.E./dl) Formen unterteilt und ist abhängig von der Art und vom Ort der Mutation: Ist beispielsweise die Promotor-Region des jeweiligen Gens verändert, wird eventuell überhaupt kein Protein gebildet, während eine Mutation innerhalb eines Exons die Aktivität beeinflussen kann. Für Faktor VIII sind mittlerweile circa 3000 und für Faktor IX mehr als 1000 Mutationen bekannt, von denen die meisten zu Missense-Austauschen führen, also zum Einbau einer anderen Aminosäure an einer bestimmten Position.
Mutationen in Genen, die für die anderen Gerinnungsfaktoren kodieren, resultieren in deutlich selteneren Koagulopathien, die autosomal rezessiv vererbt werden. Die Schweregrade der Blutungsneigungen unterscheiden sich zwischen den Patienten sehr stark, je nachdem welcher Gerinnungsfaktor betroffen und wie groß seine Restaktivität ist. Eine sehr häufige erbliche Blutungsstörung, die üblicherweise aber nicht zu den Koagulopathien gezählt wird, ist das Von-Willebrand-Syndrom, bei dem das Gen für den multimeren Von-Willebrand-Faktor (VWF) mutiert ist. Von einer schweren Form des Von-Willebrand-Syndrom sind allerdings weniger als 1 von 300.000 Menschen betroffen. VWF ist vor allem für die Interaktion zwischen Thrombozyten und der Gefäßwand wichtig und ist an der Bildung des primären weißen Thrombus beteiligt. Zudem dient VWF auch als Stabilisator für den Gerinnungsfaktor VIII, sodass es beim Von-Willebrand-Syndrom sekundär zu einem Gerinnungsdefekt kommt. Seit Kurzem ist mit Veyvondi® mit dem enthaltenen Vonicog alfa ein rekombinant hergestellter VWF auf dem Markt, der erwachsenen Patienten mit Von-Willebrand-Syndrom zur Vorbeugung und Behandlung von Blutungen eingesetzt werden kann, allerdings nicht zur Therapie einer Hämophilie A.
Lange Zeit wurde alles Mögliche ausprobiert, um Blutern das Leben zu erleichtern: Hypnose zur Ruhigstellung, Eis zur Kühlung, Sauerstoffinhalation, Schilddrüsen- und Knochenmarkpräparationen, Wasserstoffperoxid, Injektionen von Natriumcitrat, Calciumlactat oder Oxalsäure oder auch eine Therapie mit weiblichen Hormonen – schließlich waren ja Frauen praktisch nicht von der Erkrankung betroffen. Auch Bluttransfusionen wurden mit mehr oder weniger Erfolg vorgenommen, allerdings dauerte es bis 1964, bis durch die Kryopräzipitation von frisch eingefrorenem Blutplasma Faktor VIII und auch VWF und Fibrinogen aufkonzentriert werden konnten, was eine Verbesserung der Therapie brachte.
In den 1970er-Jahren standen dann tatsächlich gefriergetrocknete Konzentrate einerseits von Faktor VIII für die Therapie der Hämophilie A und andererseits der Gerinnungsfaktoren II, VII, IX und X des Prothrombin-Komplexes für die Behandlung der Hämophilie B zur Verfügung und brachten den betroffenen Patienten Erleichterung. Ein herber Rückschlag war in den 1980er-Jahren zu verzeichnen, als sich viele Hämophilie-Patienten über diese Plasmapräparate mit HIV infizierten und an Aids erkrankten. Als in den 1990er-Jahren gentechnisch hergestellte Gerinnungsfaktoren zur Verfügung standen, gab es eine neue, sichere und effiziente Therapie der Hämophilie. Ungefähr zur gleichen Zeit war ebenfalls auch die Gewinnung aus Plasma durch geeignete Virus-Abreicherungsschritte wieder sicherer geworden.
Inzwischen stehen viele verschiedene Präparate zur Verfügung, die den Patienten eine gute Lebensqualität bei normaler Lebenserwartung ermöglichen. Bei der bedarfsgerechten Verabreichung werden die Gerinnungsfaktoren je nach Schweregrad der Hämophilie und aufgetretener Blutung dosiert. Die Aktivität der Gerinnungsfaktoren wird in Internationalen Einheiten (I.E.) angegeben, die dem jeweiligen Gehalt des Proteins in einem Milliliter normalen Humanplasmas entspricht. Für die Berechnung der jeweils nötigen therapeutischen Menge an Faktorpräparat dienen die empirischen Befunde, dass 1 I.E. Faktor VIII pro kg Körpergewicht die Aktivität im Plasma um 2 I.E./dl erhöht, während 1 I.E. Faktor IX pro kg Körpergewicht die Aktivität im Plasma durchschnittlich um 0,8 I.E./dl erhöht. Zur Kontrolle der Gerinnungsaktivität im Blut wird üblicherweise die aktivierte partielle Thromboplastinzeit (aPTT) bestimmt. Allerdings zeigte sich, dass bei dieser Therapie wegen unbemerkter innerer Blutungen nach wie vor Gelenkschädigungen auftraten.
Das verbesserte sich, als die Gerinnungsfaktoren – vor allem bei den Patienten mit einer schweren, aber zuweilen auch bei solchen mit mittelschwerer Hämophilie – nicht mehr nur bei Bedarf, sondern vielmehr prophylaktisch eingesetzt wurden. Therapieziel ist dabei, die Plasmakonzentration der entsprechenden Faktoren über 1 Prozent zu halten. Dadurch wird eine gute Blutgerinnung erreicht und die Patienten leiden seltener unter Gelenkschäden. Bei Patienten mit schwerer Hämophilie A werden zur Langzeitprophylaxe zwischen 20 und 40 I.E. Faktor VIII pro kg Körpergewicht im Abstand von zwei bis drei Tagen gegeben. In manchen Fällen – besonders bei jüngeren Patienten – können eventuell höhere Dosen oder kürzere Dosierungsabstände nötig sein. Zur Langzeitprophylaxe einer schweren Hämophilie B ist die übliche Dosis einmal wöchentlich 35 bis 50 I.E. Faktor IX pro kg Körpergewicht. Gut eingestellte Patienten können mit bis zu 75 I.E. Faktor IX pro kg Körpergewicht in einem Intervall von 10 oder 14 Tagen behandelt werden.
Durch Faktorpräparate mit verlängerter Halbwertszeit (Extended Half-Life, EHL) lassen sich die Intervalle zwischen den Applikationen weiter verlängern. Zu beachten ist allerdings, dass pegylierte Gerinnungsfaktoren von der Europäischen Zulassungsbehörde EMA erst für Patienten ab zwölf Jahren zugelassen sind.
Die Blutgerinnung ist ein komplizierter und zugleich ausgeklügelter, faszinierender Vorgang, der lebensnotwendig ist. Hämophilie-Patienten haben inzwischen dank der verschiedenen verfügbaren Behandlungsmöglichkeiten eine annähernd normale Lebenserwartung. Allerdings treten nach wie vor Gelenkschäden auf. Die intravenöse Applikation der meisten Präparate bedarf einiger Übung und ist oft genug mit Komplikationen verbunden. Trotz der geringen Patientenzahlen gibt es eine Vielzahl unterschiedlicher Präparate.
Literatur
Ingram, G.I.: The history of haemophilia. J. Clin. Pathol. 29 (1976), 469–479.
Mannucci, P.M.: Hemophilia therapy: the future has begun. Haematologica. 105 (2020), 545-553.
Peters, R., Harris, T.: Advances and innovations in haemophilia treatment. Nat. Rev. Drug Discov. 17 (2018), 493-508.
Key, N.S.: Inhibitors in congenital coagulation disorders. Br. J. Haematol. 127 (2004), 379-91.
Giangrande, P.: Haemophilia B: Christmas disease.Expert Opin Pharmacother. 6 (2005), 1517-1524.
Spadarella, G., Di Minno, A., Milan, G., Franco, N., Polimeno, M., Castaldo, F., Di Minno, G.: Paradigm shift for the treatment of hereditary haemophilia: Towards precision medicine. Blood Rev. 39 (2020), 100618.
Weyand, A.C., Pipe, S.W.: New therapies for hemophilia. Blood. 133 (2019), 389-398.
Balkaransingh, P., Young, G.: Novel therapies and current clinical progress in hemophilia A. Ther Adv Hematol. 9 (2018), 49-61.


