 |  | Bettina Wick-Urban |
 |
08.12.2024 08:00 Uhr |
Wenn die Sauerstoffsättigung im Blut deutlich abfällt, brauchen Patienten mit pulmonaler arterieller Hypertonie eine externe Sauerstoffzufuhr. / © Adobe Stock/Ralf Geithe
Die pulmonale arterielle Hypertonie (PAH) ist eine relativ seltene Erkrankung. In Deutschland leiden circa 3000 bis 4000 Erwachsene daran. Bei Kindern geht man von 2 bis 2,2 Fällen pro 1 Million aus. Bei Mädchen und Frauen tritt die Erkrankung fast doppelt so häufig auf wie bei Jungen und Männern (1, 2).
Die PAH betrifft die Lungenarterien, die das Blut von der rechten Herzhälfte in die Lunge transportieren. Sie ist definiert durch einen mittleren pulmonal arteriellen Druck (PAPm) über 20 mmHg in Ruhe (normal 10 bis 11 mmHg) und einen pulmonal vaskulären Widerstand (PVR) über 2 Wood-Einheiten (WE, normal 0,7 bis 1,1). Durch den chronisch erhöhten Blutdruck in den Lungengefäßen wird das Herz in Mitleidenschaft gezogen: Der Herzmuskel verdickt sich, verliert immer mehr an Elastizität und ist letztlich nicht mehr in der Lage, das notwendige Blutvolumen zu transportieren. Es entwickelt sich eine Rechtsherzinsuffizienz. Im fortgeschrittenen Stadium kann es auch zu Veränderungen von Schilddrüse, Niere und Leber kommen (1, 3).
Die klinischen Symptome der PAH sind unspezifisch und vor allem auf die zunehmende Rechtsherzinsuffizienz zurückzuführen. Die Patienten klagen über Kurzatmigkeit bei körperlicher Belastung, Müdigkeit, körperliche Schwäche, Angina pectoris und Synkopen während und kurz nach körperlicher Anstrengung. Häufig wird die Diagnose gestellt, wenn sich der Patient von einer Atemwegsinfektion nicht vollständig erholt und das Röntgenbild ein vergrößertes Herz zeigt.
Bei Säuglingen und Kleinkindern können auch eine Trinkschwäche, rezidivierendes Erbrechen oder eine Gedeihstörung auf eine PAH hindeuten. Bei Kindern und Jugendlichen treten Entwicklungsverzögerungen auf (1, 3).
Lungenhochdruck im Kindesalter unterscheidet sich von dem der Erwachsenen insbesondere durch die spezifische Pathophysiologie der Herzfehler-assoziierten PAH, das Vorkommen von entwicklungsbedingten Lungenerkrankungen und die häufige Assoziation mit chromosomalen und genetischen Auffälligkeiten, zum Beispiel mit Trisomie 21. Vor allem bei zu früh geborenen Kindern ist PAH eine direkte Folgeerscheinung der Lungenunreife und der verzögerten Anpassung der Lunge an das postnatale Leben.
Bei circa 82 Prozent der Neugeborenen und Kinder ist die PAH transient, weil operativ behandelbar, zum Beispiel bei reparablen kardialen Shunts oder einer pulmonalen arteriolären Verengung. Bei 34 bis 49 Prozent der Neugeborenen und Kleinkinder mit nicht-transienter PAH liegt eine entwicklungsbedingte Lungenerkrankung vor, zum Beispiel eine bronchopulmonale Fehlbildung (Dysplasie), kongenitale Zwerchfellhernie oder pulmonale Gefäßanomalien (4).
Die pulmonale Vasokonstriktion wird bei Kindern – sofern nicht angeboren – und Erwachsenen durch einen Mangel an den vasodilatatorischen Botenstoffen Stickstoffmonoxid (NO) und Prostacyclin (PGI2) hervorgerufen beziehungsweise durch eine erhöhte Konzentration von Endothelin-1 (ET-1), das vasokonstriktorisch wirkt und die Fibrogenese und Zellproliferation anregt. Auch erhöhte Konzentrationen von Phosphodiesterase Typ 5 (PDE-5) werden gefunden. Das Enzym baut zyklisches Guanosinmonophosphat (cGMP) ab; dies ist ein potenter Vasodilatator, dessen Produktion durch NO stimuliert wird (5).
Das kindliche Endothelium der Lungengefäße produziert Wachstumsfaktoren, die für die Homöostase und den Gefäßtonus wichtig sind. Der Auslöser für die endotheliale Dysfunktion, die zur vermehrten Ausschüttung von vasoproliferativen Wachstumsfaktoren wie Vascular Endothelial Growth Factor (VEGF), Tumornekrosefaktor alfa (TNF-α) und Platelet Derived Growth Faktor (PDGF) sowie vasokonstriktiven Botenstoffen führt, ist noch nicht geklärt. Entzündungen und damit einhergehende erhöhte Konzentrationen von Zytokinen, Interleukinen und Chemokinen können zur Verschlechterung einer PAH beitragen. Beispielsweise können rheumatische Erkrankungen zu einer PAH-Krise führen. Bei Kindern, vor allem bei zu früh Geborenen, verschlechtern vor allem Infektionen die Symptome (5, 6).
Der Transforming Growth Factor beta-(TGF-β-)Signalweg spielt eine wichtige Rolle in der embryonalen Herzentwicklung und der Gefäßentstehung. Der Bone Morphogenetic Protein Receptor-2 (BMPR-2) ist Teil des TGF-β-Signalwegs. Dieser Rezeptor ist essenziell für die Zellhomöostase, da er die Apoptose fördert und die Proliferation hemmt. Eine Verringerung von BMPR-2 bedeutet daher ein gesteigertes Wachstum der glatten Muskelzellen und Endothelzellen in den Arteriolen. Gegenspieler des BMPR-2-Signalwegs ist der Activin-Signalweg, der vermehrt pro-proliferativ wirkt (6).
Mutationen in den für BMPR-2 und Activin kodierenden Genen (BMPR-2 oder ACVRL-1) prädisponieren für die Entstehung einer PAH im Kindesalter. Bei Trägern einer BMPR-2- oder ACVRL-1-Mutation wird meist ein aggressiverer Verlauf der Erkrankung beobachtet; die Patienten haben bereits in jüngerem Alter eine ungünstige Hämodynamik (4).
Grundsätzlich gilt für alle Patienten, die Belastung des kardiovaskulären Systems möglichst zu minimieren (Kasten).

Körperliche Schwäche und Atemnot, insbesondere unter Belastung, sind oft erste Symptome einer pulmonalen arteriellen Hypertonie. / © Adobe Stock/RFBSIP
Für Erwachsene mit PAH steht eine Reihe vasodilatatorischer Medikamente mit verschiedenen Wirkmechanismen zur Verfügung (Tabellen 1 und 2). Für Kinder und Jugendliche gibt es – neben Sildenafil und Bosentan – nun weitere zugelassene Therapieoptionen: Tadalafil, Riociguat und Ambrisentan (Tabelle 2), wodurch sich die Pro¬gnose der kleinen Patienten wesentlich verbessert hat. Während in den 1990er-Jahren das mittlere Überleben nach Diagnose bei weniger als einem Jahr lag, liegen die Fünf- und Zehn-Jahres-Überlebensraten heute bei 97 und 78 Prozent (7). Dennoch ist die PAH weiterhin eine chronisch progressive und nicht heilbare Erkrankung. Therapieziele bei Erwachsenen wie auch Kindern sind daher
Die Therapie wird individuell angepasst an den Schweregrad der Erkrankung, den klinischen Verlauf und zusätzliche Begleiterkrankungen (1, 3).
© Adobe Stock/Ralf Geithe
Wichtig bei allen Patienten ist es, die Belastung des kardiovaskulären Systems und die Erhöhung des Lungenwiderstands möglichst gering zu halten, um Synkopen (Ohnmachtsanfälle) zu vermeiden (1, 5). Daher sind folgende Maßnahmen immer wichtig:
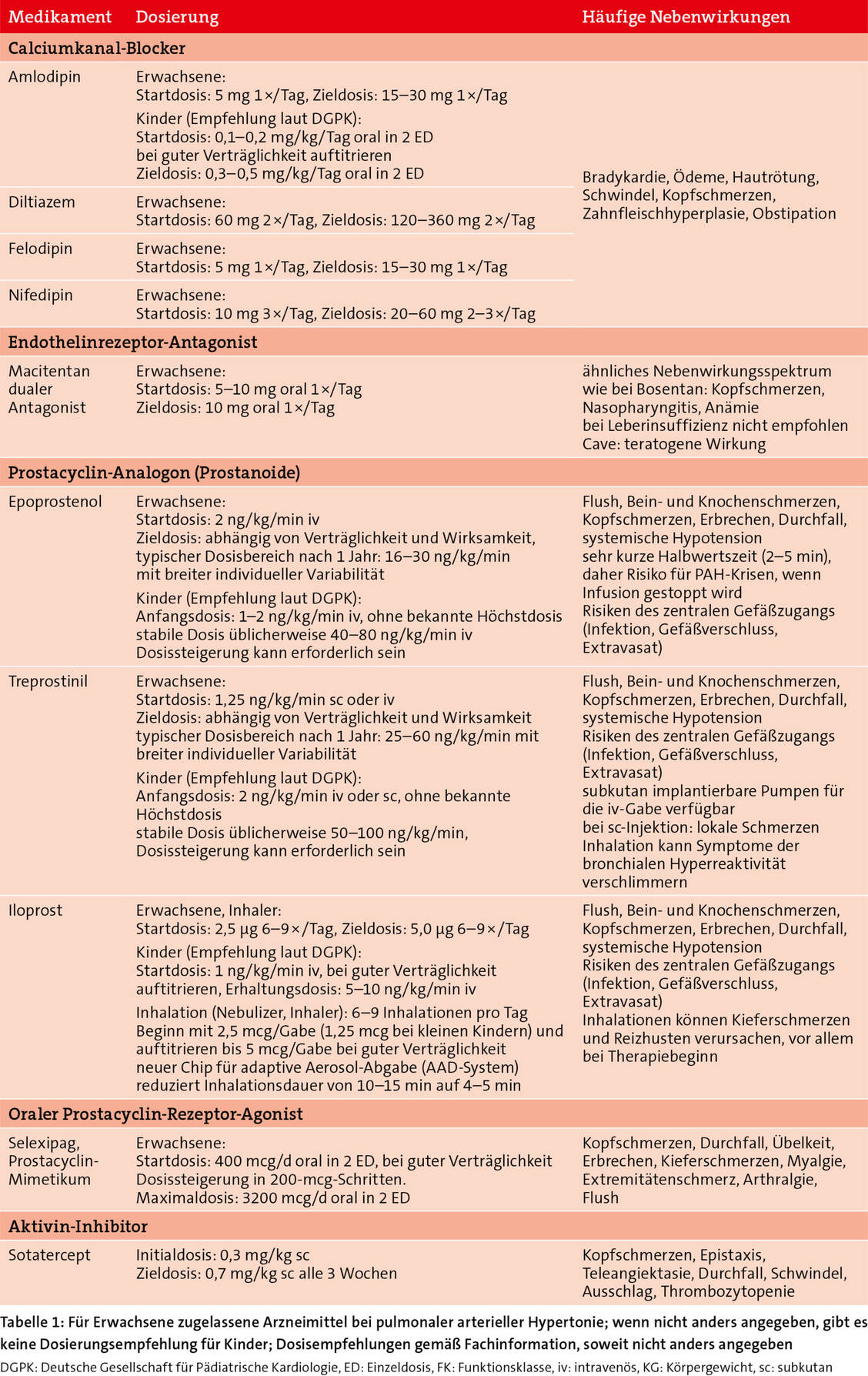
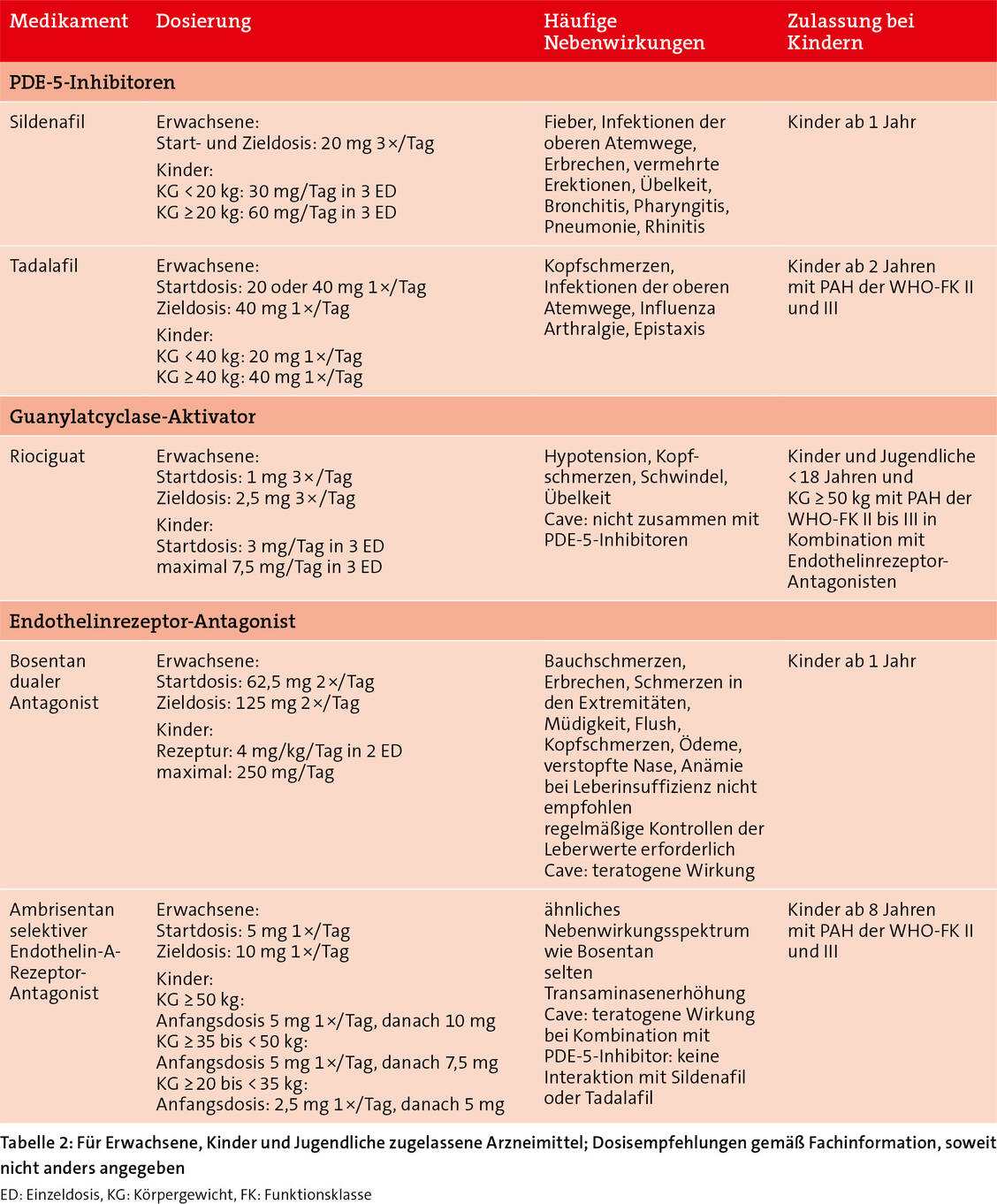
In den Anfängen der PAH-Therapie mit vasodilatatorischen Arzneimitteln galt die Lehrmeinung, dass bei Verschlechterung der Symptomatik die Therapie stufenweise eskaliert werden sollte. Aufgrund von positiven Studienergebnissen mit Kombinationstherapien wird heute bei Erwachsenen ohne kardiovaskuläre Begleiterkrankungen bereits initial eine Kombinationstherapie empfohlen.
So wurde in der AMBITION-Studie gezeigt, dass eine Kombination von Ambrisentan und Tadalafil die Zeit bis zur Verschlechterung der Erkrankung gegenüber einer Monotherapie signifikant verlängert (8). Auch scheint die Kombinationstherapie das Mortalitätsrisiko zu verringern (9).
Ähnliche Ergebnisse zeigte die TRITON-Studie für die Kombination von Macitentan und Tadalafil. Eine Triple-Therapie mit Selexipag brachte jedoch keinen zusätzlichen Vorteil (10).
Bei PAH-Patienten mit kardiovaskulären Begleiterkrankungen wird im Moment, auch mangels Studienergebnissen, immer noch eine sequenzielle Therapieeskalation empfohlen (3).
Die Kombination von Prostanoiden mit anderen PAH-Therapeutika scheint auch bei Kindern von Vorteil zu sein. So zeigte eine retrospektive Auswertung, dass sich bei den kleinen Patienten die Zeit wesentlich verlängerte, bis sie eine Lungentransplantation benötigten (11, 12). Eine randomisierte, prospektive klinische Studie (Kids Mod PAH Trial) untersucht derzeit, ob die initiale Gabe von Sildenafil plus Bosentan im Vergleich zu Sildenafil alleine einen günstigeren Effekt auf den Krankheitsverlauf hat. Erste Ergebnisse werden 2026 erwartet (13).
Während alle bislang zugelassenen Therapien für PAH eine vasodilatatorische Wirkung haben, gibt es nun auch neue Ansätze. Neue Wirkstoffe stoppen die Krankheitsprogression, indem sie Signalwege hemmen, die die Proliferation beeinflussen: Sie könnten zu einem wirklichen therapeutischen Fortschritt führen. Daneben werden neue vasodilatatorische Wirkstoffe entwickelt, die aufgrund einer verbesserten Pharmakokinetik oder einer patientenfreundlicheren Darreichungsform eine Therapieoptimierung ermöglichen.
Der PDE-5-Inhibitor Vardenafil, als Inhaler angewandt, wird entwickelt als Bedarfsmedikation bei durch Anstrengung ausgelösten Symptomen. Die inhalative Anwendung ermöglicht eine gezieltere Applikation ohne Titration und die Gabe höherer Dosen mit möglicherweise weniger systemischen Nebenwirkungen im Vergleich zu Sildenafil und Tadalafil. In einer ersten Studie verminderte die inhalative Gabe den pulmonal vaskulären Widerstand (PVR) (14) und wird derzeit weiter klinisch evaluiert.

Selbstständig gehen zu können, erhöht die Lebensqualität immens. Daher ist die Strecke, die Patienten innerhalb von sechs Minuten zurücklegen können, ein wichtiger Parameter für die klinische Wirksamkeit von PAH-Therapeutika. / © Adobe Stock/Edler von Rabenstein
Frespaciguat (MK-5475) ist ein löslicher Guanylatcyclase-Stimulator wie Riociguat, der jedoch inhalativ angewandt wird. Er zeigte in einer ersten Studie eine gute Wirksamkeit und Verträglichkeit und wird derzeit in Phase II/III klinisch erprobt (15).
Ralinepag, ein oraler, selektiver nicht-prostanoider Prostacyclin-Rezeptoragonist, wird derzeit in Phase III untersucht. Im Vergleich zu Selexipag und Treprostinil hat er eine verlängerte Halbwertszeit und stabilere Plasmaspiegel. In einer placebokontrollierten Phase-II-Studie reduzierte Ralinepag den PVR vergleichbar mit Selexipag (Tabelle 1). Die Sechs-Minuten-Gehstrecke war jedoch länger. Im Vergleich zu Placebo verschlechterte sich bei weniger Patienten unter Ralinepag die funktionelle Herzinsuffizienz-Klasse gemäß WHO/NYHA (16).
Ein Hauptziel von Arzneistoffen in der klinischen Entwicklung ist der dysregulierte TGF-β-Signalweg mit seinen Gegenspielern BMPR-2 und Activin. Dadurch erhofft man sich eine Wiederherstellung der gestörten Proliferation der glatten Muskelzellen und der Endothelzellen (6).
Ein erstes Arzneimittel, Sotatercept, wurde im Juni dieses Jahres von der europäischen Zulassungsbehörde EMA zur Anwendung in Kombination mit anderen PAH-Therapien zugelassen und kam im Oktober 2024 auf den Markt (17). Sotatercept ist ein Fusionsprotein aus der Fc-Domäne des Immunglobulins G1 und der extrazellulären Rezeptordomäne des Activin-Rezeptors IIA. Der Wirkstoff bindet als Ligandenfalle überschüssiges Activin A, das bei PAH-Patienten in erhöhter Konzentration vorliegt. In Tierversuchen verminderte die Blockade der Activin-Signalkette die vaskuläre Zellproliferation, verstärkte die Apoptose und verringerte die Entzündung in den Gefäßwänden (Tabelle 1).
In der zulassungsrelevanten Phase-III-Studie erhielten die Patienten zusätzlich zu einer stabilen Basistherapie noch Sotatercept oder Placebo alle drei Wochen subkutan injiziert. Der neue Wirkstoff verbesserte signifikant die körperliche Belastbarkeit, gemessen mit der Sechs-Minuten-Gehstrecke, um 41 Meter gegenüber Placebo sowie den pulmonal vaskulären Widerstand und die WHO-Funktionsklasse. Auch das Risiko für eine Verschlechterung der Erkrankung und das Mortalitätsrisiko sanken. Unter Sotatercept häufiger beobachtete Nebenwirkungen waren ein Anstieg des Hämoglobinwerts, Teleangiektasien, Nasen- und Zahnfleischbluten, Benommenheit, Erhöhung des Blutdrucks und Thrombozytopenien (18).
Cibotercept (KER-012) ist ein modifiziertes Activin-Rezeptor-Typ-IIB-Fusionsprotein, das selektiv als Ligandenfalle die TGF-β-Liganden Activin A, Activin B und Myostatin inhibiert; diese sind für die Proliferation der glatten Muskelzellen und die Fibrose verantwortlich. Cibotercept wird derzeit in einer Phase-II-Studie (TROPOS) an PAH-Patienten in Kombination mit einer stabilen Basistherapie untersucht (19, 20, 21).
Ein weiterer Forschungsansatz ist die Blockade des Platelet Derived Growth Factor-Rezeptors (PDGF-R). PDGF-R stimuliert die Proliferation, blockiert die Apoptose und fördert so die vaskulären Veränderungen bei PAH. Bei PAH-Patienten werden erhöhte Konzentrationen gefunden.
Imatinib, zugelassen für die chronische myeloische Leukämie, inhibiert verschiedene Tyrosinkinasen, zum Beispiel den PDGF-R auf glatten Muskelzellen. In der IMPRES-Studie verbesserte oral gegebenes Imatinib die körperliche Belastbarkeit, gemessen mit dem Sechs-Minuten-Gehtest, und reduzierte den pulmonal vaskulären Widerstand PVR. Jedoch wurde die klinische Entwicklung aufgrund von schwerwiegenden Nebenwirkungen und dadurch bedingten Studienabbrüchen gestoppt. Bei einigen Patienten, die zusätzlich mit Antikoagulanzien behandelt wurden, traten subdurale Hämatome auf (22, 23).
Inhalativ verabreichtes Imatinib (AV-101) wurde in einer Studie an gesunden Probanden gut vertragen; außer Kopfschmerzen und Husten traten keine Nebenwirkungen auf (24). Jedoch wurde die Phase-II/III-Studie im August 2024 beendet, da die Inhalation auch in der höchsten Dosis keine Wirksamkeit zeigte.

Dank intensiver Forschung sind in den letzten Jahren mehrere neue Medikamente für PAH-Patienten auf den Markt gekommen, die die Prognose deutlich verbessern. / © Adobe Stock/Westend61
Aussichtsreicher scheint Seralutinib zu sein, das den PDGF-R, den Colony Stimulating Factor-1-Rezeptor (CSF-1-R) sowie den Mast/stem Cell Growth Factor-Rezeptor kit (c-KIT) inhibiert: drei Tyrosinkinasen, die miteinander interagieren. Zudem erhöht der Wirkstoff die BMPR-2-Konzentration. Aktivierte CSF-1-R-positive Makrophagen und c-KIT-positive pulmonal arterielle Endothelzellen sezernieren PDGF, das an PDGF-R bindet, die auf den glatten Muskelzellen der Lungenarterie und Fibroblasten vorhanden sind. Die Aktivierung von PDGF-R induziert die Proliferation dieser Zellen, reguliert die Konzentration von BMPR-2 herunter und verschlimmert den proliferativen Prozess in einer sich verstärkenden Rückkopplungsschleife weiter (25). Seralutinib unterbricht diesen Prozess.
Inhalativ verabreichtes Seralutinib reduzierte in einer Phase-II-Studie (TORREY) an PAH-Patienten, die zusätzlich eine Basistherapie erhielten, signifikant den PVR gegenüber Placebo nach 24 Wochen (26). In einer unverblindeten Verlängerungsstudie verbesserte sich der Wert bei Patienten, die zuvor bereits auf die Therapie angesprochen hatten, bis zu Woche 72. Seralutinib wurde während dieser Zeit gut vertragen (27). Mit den Ergebnissen der zulassungsrelevanten Phase-III-Studie (PROSERA) wird Ende 2025 gerechnet; darin wird der Effekt von Seralutinib auf die körperliche Belastbarkeit und die Krankheitsprogression evaluiert (28).
Gentherapeutische Ansätze bei PAH sind noch weitestgehend im experimentellen Stadium. Dies liegt vor allem an den Schwierigkeiten, das genetische Material zum gewünschten Wirkort in der Lunge zu transferieren, und an der kurzen Wirksamkeit (29). In klinischen Studien, bei denen wenigen Patienten Stammzellen (Endothelvorläuferzellen) infundiert wurden, verbesserten sich die körperliche Belastbarkeit sowie hämodynamische Parameter der Patienten. Weitere Studien sind notwendig, um den Langzeiteffekt und die Nebenwirkungen besser zu verstehen (30, 31).
Ein besseres Verständnis der Pathogenese von PAH und die Verfügbarkeit von vasodilatatorischen Therapien hat bei Erwachsenen, aber vor allem bei Kindern die Prognose in den letzten Jahren wesentlich verbessert. Positive Studienergebnisse unterstützen die initiale Kombinationstherapie bei Erwachsenen und Kindern.
Neue Arzneistoffe in der klinischen Entwicklung, die das Fortschreiten der Erkrankung stoppen sollen, indem sie Signalwege hemmen, die die Proliferation beeinflussen, könnten zu einem wirklichen therapeutischen Fortschritt führen.
Bettina Wick-Urban studierte Pharmazie an der Albert-Ludwigs-Universität in Freiburg. Nach ihrer Promotion in Basel und Freiburg mit einer molekularbiologischen Arbeit im Bereich der experimentellen Onkologie arbeitete sie zunächst als Referentin bei der Arzneimittelinformationsstelle der ABDA. Danach wechselte sie in die pharmazeutische Industrie, wo sie seitdem in verschiedenen Positionen in der klinischen Forschung, Arzneimittelsicherheit und Medizin tätig ist, davon zwei Jahre in den USA. Zwischenzeitlich schloss sie ein Journalismus-Studium ab.


