 |  | Carsten Culmsee |
| | Jana Fedjaev |
| | Hanna Rosemarie Hofmann |
| | Johanna Lücke |
 |
27.10.2024 08:00 Uhr |

Mit höherem Alter steigt die Prävalenz einer Demenzerkrankung – aber viele ältere und alte Menschen sind geistig völlig fit. / © Getty Images/Maskot
Demenz gehört zu den häufigsten neurologischen Erkrankungen der älteren Menschen und ist weltweit die häufigste neurodegenerative Erkrankung überhaupt. In Deutschland sind der-zeit rund 1,84 Millionen Menschen erkrankt, davon etwa zwei Drittel mit der Verdachtsdiagnose Alzheimer-Demenz. Der Hauptrisikofaktor ist das Alter. Es handelt sich um eine altersbedingte fortschreitende Erkrankung des Gehirns, die mit milden kognitiven Einschränkungen (Mild Cognitive Impairment, MCI) beginnt und mit einer geschätzten Prävalenz der MCI etwa 3,6 bis 5,6 Millionen Menschen über 50 Jahre betrifft (1).
Die alarmierenden Prognosen für die Entwicklung der Erkrankung wurden inzwischen etwas abgeschwächt, weil die Menschen in den zunehmend überalterten Gesellschaften offenbar gesünder ein hohes Lebensalter erreichen. Es besteht aber weiterhin ein hoher Bedarf an Forschung zur Pathologie und zu effektiven Therapieoptionen. Die Möglichkeiten der Diagnostik und Therapie sind derzeit überschaubar. Eine kausale Therapie, um Demenz nachhaltig aufzuhalten oder gar zu heilen, gibt es nicht.
Das gilt insbesondere für die Alzheimer-Demenz, mit circa 60 Prozent die häufigste Form der Demenzen. Proteinablagerungen im Gehirn, die Amyloid-Plaques, gelten seit der Erstbeschreibung durch Alois Alzheimer vor etwa 120 Jahren als das charakteristische Kennzeichen dieser Demenzform. Sie kann schon in sehr frühem Lebensalter (um die 40) auftreten, schreitet rasch fort und führt innerhalb von etwa zehn Jahren nach Diagnosestellung zum Tod (1, 2).
Charakteristisch sind daneben Anreicherungen von Tau-Fibrillen (Neurofibrillen) in den degenerierten Neuronen, die zunehmend funktionelle und strukturelle Schädigungen aufweisen. In der Folge schneiden Patienten in Gedächtnis- und Kognitionstests immer schlechter ab. Das Gehirn zeigt immer geringere Stoffwechselaktivität und verliert auch an Masse: Es schrumpft geradezu (2, 3).
Lange vor der Manifestation der Gedächtnis- und Verhaltensstörungen kommt es zu Veränderungen im Gehirn mit zunehmenden Ablagerungen der Amyloid-Plaques, entzündlichen Prozessen, Stoffwechselveränderungen und der Ausbildung der Tau-Neurofibrillen (Abbildung 1) (4). Die klinische Diagnosestellung kommt also sehr spät, wenn die Degeneration schon weit fortgeschritten ist.
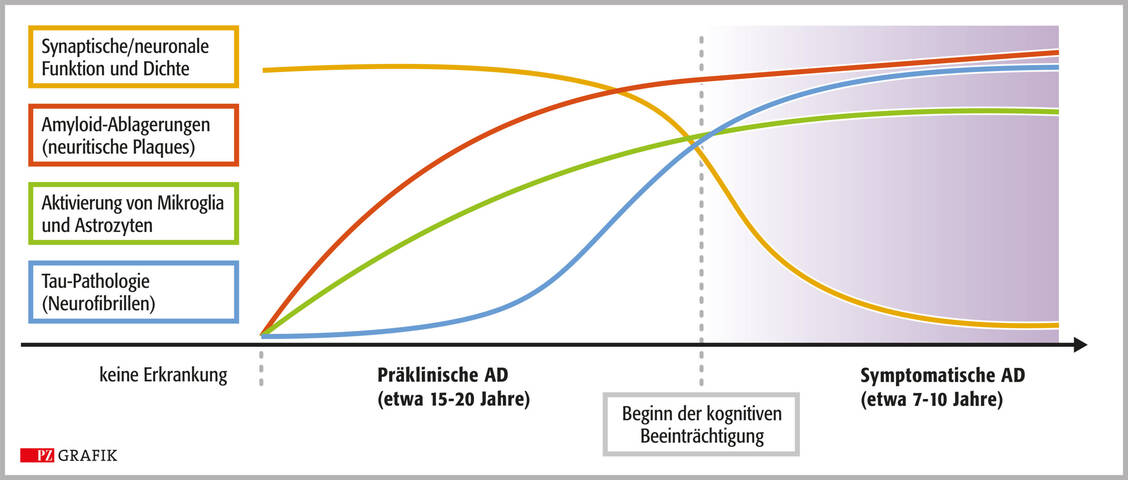
Abbildung 1: Pathologische und klinische Veränderungen im jahrzehntelangen Verlauf der Alzheimer-Demenz (AD); modifiziert nach: DOI 10.1016/j.cell.2019.09.001 / © PZ/Stephan Spitzer
Die aktuelle Therapie der Demenz ist überschaubar. Mit Ginkgo-Präparaten kann bei vaskulärer Demenz oder milder kognitiver Einschränkung ein Therapieversuch gestartet werden. Es kann zu Verbesserungen der Alltagskompetenzen und im Gesamteindruck der Demenzkranken kommen; eine signifikante Verbesserung kognitiver Leistungen ist aber klinisch nicht nachzuweisen (5).
Die weitere Pharmakotherapie umfasst die Acetylcholinesterase-(AChE-)Hemmer Donepezil, Rivastigmin und Galantamin sowie bei schweren Stadien auch den NMDA-Rezeptorblocker Memantin. Tatsächlich lassen sich damit Verbesserungen von kognitiven Leistungen erreichen; Alltagskompetenzen und der Gesamteindruck werden verbessert. Allerdings können die Wirkstoffe die Neurodegeneration nicht aufhalten und die Demenz schreitet trotz Pharmakotherapie weiter fort (5).
Da es sich um Wirkstoffe mit ausgeprägtem Potenzial für unerwünschte Nebenwirkungen handelt, ist die Langzeitgabe bei fortschreitender Demenz und zunehmendem Auftreten unerwünschter Wirkungen immer wieder auf den Prüfstand zu stellen. Hier gab es aber auch positive Nachrichten: AChE-Hemmstoffe können das Langzeitüberleben offenbar günstig beeinflussen und die Patienten können von Kombinationen der Wirkstoffe profitieren (6, 7).
Aus der Forschung sind genetische Risikofaktoren für Morbus Alzheimer bekannt, zum Beispiel sehr seltene Mutationen im Gen für das Amyloid-Vorläuferprotein oder im Presenilin-Gen, die früh zu massiven Amyloid-Ablagerungen im Gehirn und zu rasch fortschreitender Alzheimer-Demenz weit vor dem 60. Lebensjahr führen können (2).
Amyloid-beta (Aβ) wird im Körper fortwährend in geringen Mengen aus einer Vorstufe, dem Amyloid-Precursor-Protein (APP), gebildet. Der Hauptteil des APP wird über die Aktivität von Alfa-Sekretasen zu einem löslichen (soluble) sAPP umgesetzt, das unter anderem neurotrophe Eigenschaften hat und neuronale Funktionen günstig beeinflussen kann. Durch die Aktivität von Beta-(BACE-) und Gamma-Sekretasen kommt es aber auch zur Bildung kleinerer Amyloid-Fragmente, Aβ40 und Aβ42, die neurotoxische, wenig lösliche Oligomere bilden und sich in den Amyloid-Plaques ablagern (8).
Die physiologische Funktion der Amyloid-Fragmente ist ebenso umstritten wie die Amyloid-Hypothese der Alzheimer-Demenz selbst (9, 10). Es gibt Hinweise darauf, dass geringste Konzentrationen der Aβ-Fragmente die neuronale Plastizität und Hirnfunktionen sogar eher verbessern und erst die übermäßige Bildung der Fragmente und der Oligomere die bekannten neurotoxischen Effekte vermittelt (9, 11). Allerdings wurden auch bei gesunden älteren Menschen im Gehirn Amyloid-Plaques nachgewiesen, ohne dass Zeichen einer Demenz erkennbar waren (12, 13).
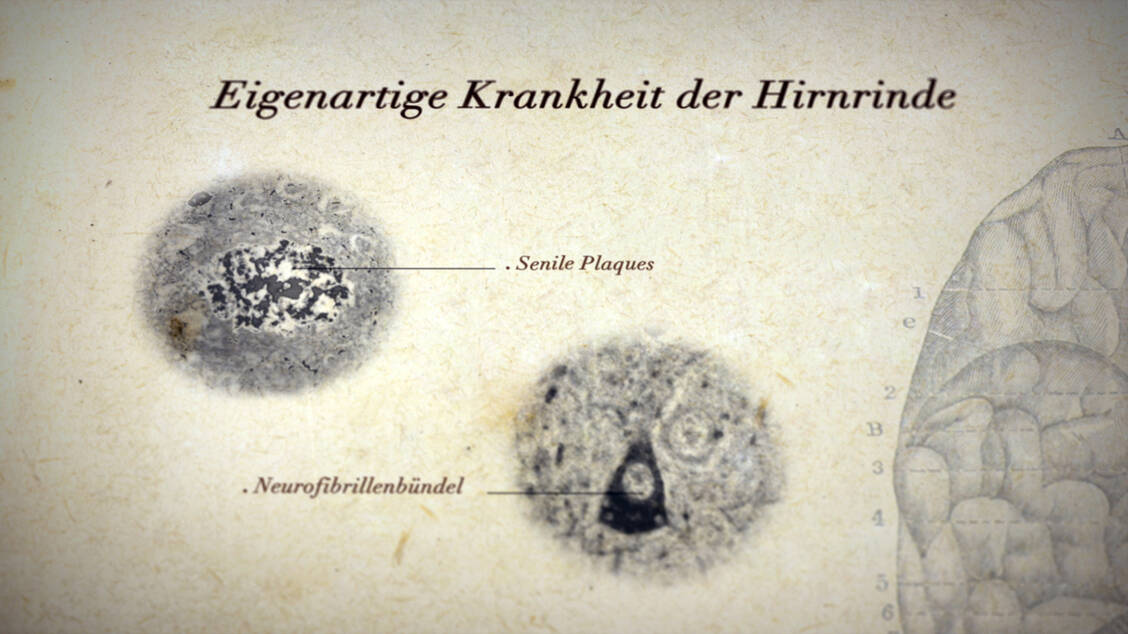
Amyloid-Plaques und Neurofibrillen-Bündel: Der deutsche Neurologe Alois Alzheimer beschrieb 1906 als Erster die charakteristischen Veränderungen im Gehirn seiner verstorbenen Patientin Auguste Deter. / © Alzheimer Forschung Initiative
Inzwischen gilt die Hypothese der toxischen Amyloid-Oligomere, die als lösliche Neurotoxine das Gehirn durchdringen sollen, zumindest als umstritten, auch weil einige der Schlüsselarbeiten nicht reproduzierbar waren (9, 14, 15). Zudem waren pharmakologische Ansätze, die in klinischen Studien auf eine Aktivierung der Alfa-Sekretasen zur bevorzugten Bildung von sAPP abzielten, ebenso wenig erfolgreich wie Studien mit BACE-Inhibitoren oder Inhibitoren der Gamma-Sekretasen zur Unterdrückung der Aβ-Produktion (16).
Im Gegensatz zum Einsatz in Tiermodellen der Alzheimer-Demenz sind alle Studien am Menschen mit den Sekretase-Modulatoren bislang negativ verlaufen. Kein Wirkstoff hat je Zulassungsstatus erreicht. Limitierend sind offenbar die zu späte Anwendung bei fortgeschrittener Demenzerkrankung oder eine eingeschränkte Dosierung aufgrund von Nebenwirkungen (17, 18).
Das Apoε4-Allel, das für ein Apolipoprotein codiert und bei familiären Alzheimer-Formen gefunden wird, bestimmt das Risiko der Erkrankung wesentlich mit. Inzwischen wird die Apoε4-Mutation als eigenständige genetische Erkrankung definiert, da homozygote Träger der Mutation an einer besonderen Form der Alzheimer-Demenz erkranken (19, 20). Dies passiert etwa zehn Jahre früher als bei Trägern des protektiven Apoε3-Allels (21, 22). Träger des Apoε2-Allels sind gegen Alzheimer geschützt (23, 24).
Darüber hinaus wurden weitere genetische Faktoren identifiziert, die jeweils für sich gesehen ein relativ geringes Risiko für eine Demenzerkrankung mit sich bringen und erst im Zusammenhang mit Umwelteinflüssen und dem Lebensstil die Entwicklung der Alzheimer-Demenz begünstigen (2, 4, 25).
Unter den neueren Risikogenen sind möglicherweise solche für zukünftige Therapieansätze interessant, die in die Regulation von Entzündungsprozessen im Gehirn eingreifen, zum Beispiel TREM2 (Triggering Receptor expressed on Myeloid cells 2) (26–28). Die Alzheimer-Erkrankung schreitet bei Patienten mit hohem TREM2-Spiegel langsamer voran. Das Protein gilt auch als möglicher diagnostischer Marker, da seine Konzentration im Liquor in der Frühphase der Erkrankung ansteigt. Mutationen im TREM2-Gen sind mit einem erhöhten Risiko für die Alzheimer-Demenz assoziiert. Hier zeigt sich die enge Verbindung der Amyloid- und Tau-Pathologie mit erhöhter Entzündungsaktivität im Gehirn (Abbildung 1) (29–33).
Die proinflammatorisch aktivierte Mikroglia ist ebenso wie die erhöhte Amyloid-Last und die Tau-Hyperphosphorylierung im Gehirngewebe bereits lange vor den klinischen Demenzsymptomen nachweisbar. Die Mikroglia verstärkt offenbar über Entzündungsreaktionen die Amyloid-Toxizität und die neuronale Degeneration (26–28, 34). Eine Hemmung der entzündlichen Mikroglia-Aktivierung hin zu einer eher protektiven Funktion dieser immunologischen Wächterzellen im Gehirn gilt als mögliche Strategie für künftige therapeutische Interventionen in der Frühphase der Erkrankung (30, 34, 35). Bislang sind Forschungserfolge vor allem im präklinisch-experimentellen Bereich zu verzeichnen; ein Durchbruch bei klinischen Studien ist derzeit noch nicht absehbar (34, 36, 37).
In jüngerer Zeit haben Antikörperstudien (wieder) aufhorchen lassen (38, 39). Eine Antikörpertherapie gegen das Amyloid-Protein scheint greifbar nahe. Die Idee ist ebenso genial wie einfach: Spezifische Antikörper sollen die Amyloid-Plaques abräumen und lösliche Amyloid-Oligomere aus dem Gehirn entfernen (40). Frühere Antikörperstudien waren teils erfolglos oder wurden wegen bedeutender Sicherheitssignale abgebrochen (41).
Mit Aducanumab wurde erstmals ein Anti-Amyloid-Antikörper zur Behandlung der Alzheimer-Demenz von der U.S. Food and Drug Administration (FDA) zugelassen. Er kam allerdings nicht auf den Markt, weil schon die Zulassung gegen den Rat des Expertengremiums erfolgte und die Ergebnisse der Zulassungsstudien unter Neurologen sehr kontrovers beurteilt wurden. Die klinischen Studien zeigten zwar einen Abbau der Amyloid-Plaques (42, 43), jedoch hatte der Antikörper keine Auswirkungen auf die Verschlechterung der kognitiven Parameter bei den Patienten (44, 45).
Zudem zeigten sich teilweise ausgeprägte Nebenwirkungen, zum Beispiel Hirnschwellungen, Wassereinlagerungen im Hirngewebe sowie Übelkeit und Schwindel. ARIA (Amyloid-related imaging abnormalities) traten bei Patienten mit schnellem Verlauf schon vor Erreichen der Zieldosis von 10 mg/kg auf. Diese Ödeme oder Mikroblutungen im Gehirn erfordern eine engmaschige Überwachung der Patienten unter der Therapie mit Amyloid-Antikörpern (Kasten) (46–48).
Die Europäische Arzneimittelagentur (EMA) hat für Europa die Zulassung für Aducanumab folgerichtig abgelehnt. Dennoch wurden hier Entwicklungen für Antikörpertherapien angestoßen, die ebenso wie die Anwendung von Gantenerumab, Donanemab oder Lecanemab wichtige Erkenntnisse für die Amyloid-Hypothese und den klinischen Nutzen dieser Therapiestrategie lieferten (39, 49–52).
Werden monoklonale Antikörper gegen Amyloid-beta therapeutisch eingesetzt, müssen die Patienten vor allem in der Frühphase der Behandlung überwacht werden, da unerwünschte Nebenwirkungen wie ARIA auftreten können. ARIA steht für Amyloid-related imaging abnormalities.
In klinischen Studien wurden bei manchen Patienten Anomalien in MRT-Aufnahmen des Gehirns gefunden, die auf vasogene Ödeme (ARIA-E) oder zerebrale Mikroblutungen (ARIA-H) hinweisen können. Diese ARIA traten weitgehend symptomfrei auf. Bei einigen Patienten kam es aber auch zu Kopfschmerzen, Übelkeit oder Schwindel; bei wenigen Patienten traten Hirnblutungen auf. Die Behandlung mit Amyloid-Antikörpern erfordert daher eine regelmäßige MRT-Überwachung, um beim Auftreten von ARIA die Therapie zumindest zu unterbrechen.
Die Wirkweise des IgG-Antikörpers ähnelt einer passiven Immunisierung, sodass die toxischen Amyloid-Fragmente neutralisiert und Amyloid-Plaques abgebaut werden sollten. In den Jahren 2014 und 2015 wurden die ersten Studien abgebrochen, da es keine Aussichten auf ein positives Ergebnis gab.
Im Jahr 2019 starteten die Phase-III-Studien GRADUATE-1 und -2, in denen eine höhere Dosis an Gantenerumab über einen Zeitraum von 27 Monaten an circa 2000 Patienten angewendet wurde. Insgesamt gab es nur geringe und nicht-signifikante Effekte. Auch in einer weiteren Studie zu dominant vererbter Alzheimer-Krankheit (DIAD) zeigte Gantenerumab keinen positiven Einfluss auf die Verlangsamung des kognitiven Verfalls bei den Patienten nach einer Behandlung über vier bis sieben Jahre.
Die klinischen Studien gelten als gescheitert und Gantenerumab hat die Beantragung zur Zulassung nicht erreicht.
Dagegen erhielt der monoklonale IgG1-Antikörper Lecanemab im Januar 2023 unter dem Handelsnamen Leqembi™ eine Zulassung in den USA. Er hatte in einer Phase-III-Studie Ende 2022 positive Ergebnisse geliefert (49).
In der EU wurde im Februar 2023 eine Zulassung beantragt. Der Ausschuss für Humanarzneimittel der EMA hat sich aber kürzlich gegen die Zulassung von Lecanemab ausgesprochen. Wesentlicher Grund waren laut EMA die vergleichsweise geringen klinischen Effekte auf den kognitiven Verfall in Relation zum Risiko möglicher schwerwiegender Nebenwirkungen. Solche Nebenwirkungen sind beispielsweise die ARIA mit ihren Auffälligkeiten in MRT-Aufnahmen des Gehirns (53). In einigen Fällen sind auch schwerwiegende klinische Störungen und Hirnblutungen aufgetreten, sodass die Therapie mit Lecanemab regelmäßig teure bildgebende Verfahren zur Überwachung von ARIA erfordert. Dies wiegt den moderaten Nutzen laut EMA nicht auf.

Etwas vergessen? Die neuen Antikörperpräparate eignen sich weder zur Prophylaxe einer Demenzerkrankung noch zur Behandlung von fortgeschrittenen Stadien. / © Adobe Stock/contrastwerkstatt
Getestet wurde Lecanemab in einer multizentrischen Phase-III-Studie an 1795 Patienten in einer frühen Phase der Alzheimer-Erkrankung im Alter von 50 bis 90 Jahren. Die Patienten erhielten Infusionen mit Lecanemab (10 mg/kg Körpergewicht) oder Placebo alle zwei Wochen über eine Laufzeit von 18 Monaten. Der primäre Endpunkt war die Änderung der Punktzahl auf der CDR-SB-Skala nach 18 Monaten. In der Lecanemab-Gruppe zeigten sich Veränderungen auf der Demenz-Skala um 1,21 Punkte und in der Placebogruppe um 1,66 Punkte (49). Der Behandlungseffekt war statistisch signifikant; die Reduktion des kognitiven Verfalls um absolut 0,45 Punkte ist jedoch klinisch wenig relevant.
Eine Teilstudie an 698 Patienten ergab zudem, dass die Aβ-Last im Gehirn unter Lecanemab signifikant verringert wurde. Damit kann diese Studie als erster klinischer Nachweis der Amyloid-Hypothese bewertet werden, da sowohl eine signifikante Reduktion der Amyloid-Last als auch eine signifikante Beeinflussung der kognitiven Verschlechterung nachgewiesen wurden (49).
Der Antikörper Donanemab greift nicht wie die anderen Antikörper am Aβ selbst an, sondern am Aβ-Protein-N3-Pyroglutamat (N3pG). Dies ist eine besonders toxische und aggressive Form des Amyloid-Peptids, der eine Katalysatorfunktion bei der Bildung von Amyloid-Plaques zugeordnet wird und die insbesondere die neurotoxischen Effekte von Aβ und hyperphosphoryliertem Tau potenziert (54).
Donanemab wurde in einer Phase-II-Studie mit 257 Patienten in einem frühen Alzheimer-Stadium und Zeichen von Tau- und Amyloid-Ablagerungen in Positronen-Emissions-Tomografie-Untersuchungen (PET) getestet (51). Die Patienten erhielten intravenös Donanemab (700 mg für die ersten drei Dosen, danach 1400 mg) oder Placebo jeweils im Abstand von vier Wochen. Der primäre Endpunkt war eine Veränderung auf der Integrated Alzheimer’s Disease Rating Scale (iADRS), die Kognition und Alltagsfunktionen erfasst, nach 76 Wochen.
Die Auswertung ergab einen positiven Einfluss von Donanemab auf den iADRS-Score, der sich unter Verum um 6,86 und in der Placebogruppe um 10,06 Punkte verschlechterte. Sekundäre Endpunkte bezogen sich auf weitere Skalen, die ebenfalls Kognition und Alltagsfunktionen bewerten. Hier zeigten sich keine signifikanten Effekte von Donanemab gegenüber Placebo (51). Der Antikörper kann demnach die kognitiven Funktionen nicht vollständig erhalten, aber den Krankheitsverlauf signifikant abbremsen. Auch die Amyloid-Belastung im Gehirn war deutlich geringer als in der Placebogruppe (50, 52).
In der randomisierten, doppelblinden, multizentrischen und placebokontrollierten Phase-III-Studie TRAILBLAZER-ALZ 2 wurde Donanemab bei 1734 Alzheimer-Patienten im Alter von 65 bis 85 Jahren getestet (50). 552 Teilnehmer hatten eine stark ausgeprägte Tau-Pathologie, während bei den restlichen 1182 Studienteilnehmern eine niedrige oder mittelgradige Tau-Pathologie im frühen Krankheitsstadium und eher schwache Demenzsymptome vorlagen. Die vierwöchentlichen intravenösen Injektionen wurden nicht über die gesamte Studiendauer von 76 Wochen fortgeführt, sondern dann abgesetzt, wenn die Amyloid-Plaque-Menge der Studienteilnehmer auf einen gewissen Wert abgesunken war. Mittels PET-Imaging wurde gemessen, dass die Plaques nach sechs Monaten bei 34 Prozent und nach zwölf Monaten bei 71 Prozent der Patienten auf einen Wert absanken, der als Alzheimer-negativ betrachtet werden kann.
Wie in der Phase-II-Studie wurden auch hier die iADRS- und CDR-SB-Skalen zur Quantifizierung der Symptome und als primäre Endpunkte verwendet. Donanemab konnte die kognitive und funktionelle Verschlechterung bei den 1182 Patienten mit mäßiger Tau-Pathologie nach 18 Monaten im Vergleich zu Placebo signifikant um 35 Prozent (p
Die positiven Outcomes der Studie veranlassten die FDA, Donanemab unter dem Handelsnamen Kisunla™ in den USA für die Behandlung von frühsymptomatischer Alzheimer-Krankheit zuzulassen. Das Pharmaunternehmen Eli Lilly stellte auch einen Zulassungsantrag an die EMA – dieser wird derzeit geprüft.

An neuen Zielstrukturen für die Demenztherapie und möglichen Liganden wird intensiv geforscht. / © Alzheimer Forschung Initiative/RTHW
Schwerwiegende Nebenwirkungen, insbesondere ARIA, die als Klassenerscheinung der Anti-Amyloid-Antikörper gelten, sind im Blick zu behalten. In der Studie kam es zu drei Todesfällen in der Verumgruppe und einem in der Placebogruppe (50). Es wurde auch festgestellt, dass insgesamt das Hirnvolumen unter Donanemab abnimmt und sich die Hirnventrikel vergrößern (51, 55).
Eine Metaanalyse legt nahe, dass auch andere Anti-Amyloid-Antikörper das Gehirnvolumen vermindern und die Ventrikel vergrößern (55). Das Gehirn schrumpft offenbar unter der Therapie, allerdings nicht in der Hippocampusregion im Schläfenlappen, sodass kognitive Funktionen anscheinend nicht betroffen sind oder dennoch günstig beeinflusst werden.
Zusammenfassend ist festzuhalten, dass die Ergebnisse der klinischen Studien mit Donanemab und Lecanemab grundsätzlich Hoffnung auf eine innovative Antikörper-gestützte Behandlung bei Demenz geben. Auch wenn die klinische Relevanz der Effekte eher gering ist, die Applikationsintervalle von zwei bis vier Wochen über Jahre hinweg ungünstig erscheinen und schwerwiegende Nebenwirkungen wie ARIA zu beachten und zu überwachen sind: Erstmals ist bei Patienten der klinische Nachweis gelungen, dass Aβ das Fortschreiten der Alzheimer-Erkrankung mitbestimmt und als therapeutischer Angriffspunkt relevant ist.
Allerdings müssen die Antikörper in einem sehr frühen Stadium der Alzheimer-Erkrankung eingesetzt werden, also bei Patienten mit gering eingeschränkter kognitiver Leistungsfähigkeit, bevor die Neurodegeneration zu weit fortgeschritten ist. Die Optimierung und Überwachung der Therapie sowie die Erkennung von geeigneten Patienten sind wesentliche Herausforderungen für den künftigen Erfolg dieses Ansatzes.
In weiteren Studien wurde bei Patienten die Bildung von Auto-Antikörpern gegen das Aβ-Protein nachgewiesen. Aβ pharmakologisch frühzeitig zu binden, könnte möglicherweise Vorteile gegenüber den biotechnologisch hergestellten Antikörpern bieten (56–58). Einer Arbeitsgruppe der Technischen Universität München gelang es in einem Beta-Amyloidose-Mausmodell, die löslichen Vorläufer der Amyloid-Plaques, unter anderem Aβ-Monomere oder -Oligomere, mithilfe von sogenannten Anticalinen abzufangen.
Anticaline sind synthetisch hergestellte, kleine Proteine, die auch niedermolekulare Antigene spezifisch binden können, sodass das Prinzip dem eines Antikörpers ähnelt. Allerdings sind Anticaline weniger immunogen und können besser in Gewebe eindringen. Das Aβ-bindende Anticalin konnte im Mausmodell die Aggregation von neurotoxischen Aβ-Oligomeren unterbinden und somit früh auftretende neuronale und synaptische Dysfunktionen umkehren (59).
Andere Strategien richten sich gegen das Tau-Protein, das in Form der Neurofibrillen ebenfalls zur Neurodegeneration beiträgt.
Beispielsweise wurde Semorinemab, ein IgG4-Antikörper, der die N-terminale Domäne des Tau-Proteins bindet, in einer Phase-II-Studie an 457 Patienten im Alter zwischen 50 und 80 Jahren mit beginnender bis milder Alzheimer-Demenz getestet. Sowohl in der Verum- als auch in der Placebogruppe wurde kein Einfluss auf die Verschlechterung der kognitiven Fähigkeiten festgestellt. Liquor-Analysen zeigten zwar, dass pTau-Werte unter Semorinemab geringfügig abnahmen, jedoch konnten in PET-Untersuchungen keine signifikanten Unterschiede hinsichtlich zerebraler Atrophie oder Tau-Ablagerung nachgewiesen werden (60).
Ob die Langzeitanwendung solcher Antikörper ausreichend sicher ist und zumindest für einen Teil der Demenzpatienten in frühen Erkrankungsphasen einen Nutzen bezüglich der kognitiven Funktionen bringt, müssen weitere klinische Studien erst noch zeigen.
Aber auch die Amyloid-Antikörper könnten sich auf die Tau-Pathologie auswirken. In der Phase-II-Studie mit Donanemab wurden neben den kognitiven Fähigkeiten auch die Tau-Spiegel über PET-Screening bestimmt. Im Vergleich zu Placebo wurde eine signifikante Verlangsamung der gesamten Tau-Akkumulation um 34 Prozent erreicht, vor allem im Schläfen-, Scheitel- und Frontallappen des Großhirns, also genau den Regionen, die von der Alzheimer-Demenz stark betroffen sind. Die Tau-Akkumulation war besonders bei den Patienten verringert, bei denen auch eine vollständige Amyloid-Plaque-Clearance erreicht wurde. In manchen frontalen Hirnregionen wurde das Fortschreiten der Tau-Agglomeration beinahe komplett gestoppt. Die Reduktion der Amyloid- und der Tau-Spiegel korrelierte insbesondere dann, wenn der Effekt früh nach Behandlungsbeginn eintrat.
Die Ergebnisse der Studie deuten darauf hin, dass die Änderung der Amyloid-Spiegel auch für das Fortschreiten der Tau-Pathologie relevant sein könnte.
Neben den Amyloid-Antikörpern mit indirekten Effekten auf die Tau-Pathologie werden derzeit auch humanisierte monoklonale Anti-Tau-Antikörper erforscht. Dies gilt ebenso für Effekte über Translationsinhibition durch den niedermolekularen Wirkstoff Buntanetap, mesenchymale Stromazellen (Lomecel-B) sowie Repurposing des für die AD(H)S-Therapie zugelassenen Noradrenalin-Wiederaufnahme-Hemmers Atomoxetin. Studien zeigen bislang weitgehend positive Resultate. Dabei handelt es sich jedoch um Phase-I- und -II-Studien, und erst klinische Studien der Phase III können klinisch relevante Nachweise der Wirksamkeit liefern.
Für die bisher bekannten therapeutischen Ansätze ist es wichtig, dass die Alzheimer-Demenz frühzeitig erkannt wird. Bildgebende Verfahren mittels PET oder MRT, Liquor- und genetische Untersuchungen, aber auch neuere und relativ einfache Blutanalysen ermöglichen inzwischen eine frühe Diagnosestellung. Dies eröffnet die Option, Patienten frühzeitig prophylaktischen oder therapeutischen Maßnahmen zuzuführen.
Wenn Patienten Symptome einer MCI aufweisen oder solche, die auf eine Alzheimer-Demenz hindeuten, kann in Blutproben auf Biomarker wie Aβ42, Aβ40 und pTau217 getestet werden. Diese gelten als charakteristisch für eine Alzheimer-Erkrankung und ermöglichen eine Abschätzung der pathologischen Situation des Patienten, die dann die weitere Diagnostik und Therapie mitbestimmt (61).
Bei einem Biomarker-Ergebnis, das als mittleres Risiko oder als »Grauzone« eingestuft wird, folgen Liquor- oder PET-Analysen. Für den Nachweis von Aβ-Plaques und Tau-Neurofibrillen mittels PET-Imaging gibt es verschiedene Radiopharmazeutika wie 18F-Florbetaben oder 18F-Flortaucipir, die das Blutprobenergebnis bestätigen können. Die Aβ- und Tau-Werte aus Liquoranalysen korrelieren stark mit denen der PET-Messungen, sodass nicht beide Methoden an demselben Patienten erforderlich sind.
Mit dieser Stratifizierung und der bestätigten Alzheimer-Diagnose kann anschließend die adäquate Pharmakotherapie gestartet werden (Abbildung 2) (62–64).
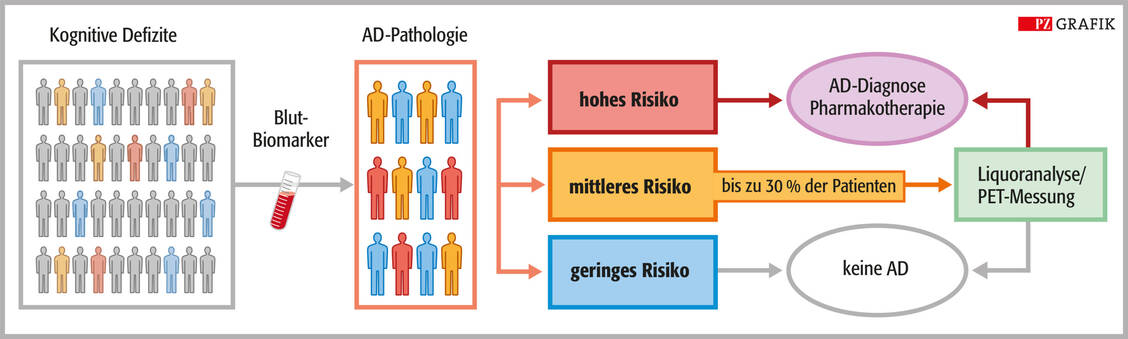
Abbildung 2: Schema einer Biomarker-basierten Alzheimer-Diagnostik. Patienten mit kognitiven Defiziten können auf Alzheimer-typische Biomarker im Blut getestet werden. Anhand des sich daraus ergebenden Risikos kann eine Alzheimer-Demenz (AD) ausgeschlossen oder durch eine anschließende Liquor-/PET-Analyse bestätigt werden. Basierend auf den Analysenergebnissen kann gegebenenfalls eine (Antikörper-)Pharmakotherapie eingeleitet werden. Modifiziert nach: DOI 10.1038/s43587-023-00403-3 / © PZ/Stephan Spitzer
Zudem muss eine bessere Differenzierung der Patientengruppen erfolgen, zum Beispiel über eine Stratifizierung der genetischen Risikofaktoren. So besteht bei Trägern des Risikogens Apoε4 ein signifikanter Zusammenhang zwischen der Amyloid-Plaque-Reduktion und dem Effekt auf die kognitiven Fähigkeiten (65). Pharmakogenomische Untersuchungen könnten also die Patienten identifizieren, die besonders gut auf die Amyloid-Antikörpertherapie ansprechen sollten – allerdings auch ein erhöhtes Risiko für ARIA mitbringen (65, 66).
Auch die vielfältigen genetischen und Umweltrisiken der Alzheimer-Demenz selbst müssen weiter erforscht werden, da immer noch nicht geklärt ist, wie es genau zum Ausbruch und Fortschreiten der Erkrankung kommt. Viele Risiken können durch Lebensstilmaßnahmen abgemildert werden (25). Physiologische (Sehschwäche, Hörverlust), psychische (Depressionen) oder externe Bedingungen wie soziale Isolation können – vor allem kombiniert – zu einer kognitiven Verarmung führen. Fehlen längerfristig wichtige »Inputs«, die man sonst durch Lesen, Diskutieren, Unterhaltung oder kognitive Herausforderungen erlangt, steigt das Alzheimer-Risiko bedeutend.
Neben der beeinträchtigten kognitiven Stimulierung zählen auch körperliche Erkrankungen (metabolisches Syndrom) sowie Rauchen und übermäßiger Alkoholkonsum zu den größten Risiken für die Entstehung der Alzheimer-Demenz (Kasten). Auch die Vermeidung von Infektionen durch Impfung kann das Demenzrisiko beeinflussen (67); tatsächlich verschlechtern Erkrankungen wie Grippe, Covid-19 oder Herpes zoster nicht nur akut die Demenzsymptomatik, sondern begünstigen auch die weitere kognitive Verschlechterung und erhöhen die Mortalität.

© Getty Images/ljubaphoto
Die Lancet-Kommission zur Prävention, Intervention und Pflege von Demenz hat kürzlich in einer Studie die Zahl der Demenzrisikofaktoren auf 14 erhöht (DOI: 10.1016/S0140-6736(24)01296-0). Neu hinzugekommen sind abnehmendes Sehvermögen und zu hohe LDL-Cholesterolwerte. Die Risikofaktoren im Überblick:
Dass Maßnahmen zur Risikominderung und Frühinterventionen durchaus signifikante Effekte bringen können, belegen jüngste Erkenntnisse aus genetischen Studien. In Familien mit Presenilin-Mutationen, die in aller Regel und unvermeidlich zu einem frühen Ausbruch der Alzheimer-Demenz führen, wurden Menschen identifiziert, die trotz der Mutation nicht erkranken (68). Auch die Ernährung hat einen wesentlichen Einfluss auf die Demenzentwicklung und zwar unabhängig vom genetischen Risiko, wie Studien zu Effekten der mediterranen Diät immer wieder belegen (69).
Solche präventiven Maßnahmen und daraus abgeleitete schützende Mechanismen sind wegweisend für künftige effektive Therapieansätze zur Prophylaxe und Behandlung von Alzheimer-Patienten (70).
Carsten Culmsee studierte Pharmazie in Marburg und wurde dort 1997 am Institut für Pharmakologie und Toxikologie promoviert. Nach einem Postdoc-Aufenthalt mit DFG-Stipendium am Sanders Brown Research Center on Aging an der University of Kentucky, Lexington, USA, einer Tätigkeit als Gruppenleiter und Dozent an der Universität Marburg (2000 bis 2003) und am Zentrum für Arzneimittelforschung der Universität München (bis 2007) kehrte er 2007 als Professor für Klinische Pharmazie an die Universität Marburg zurück. Zudem ist er Prodekan des Fachbereichs Pharmazie.
Jana Fedjaev studierte Pharmazie an der Universität Münster. Nach einem Forschungsaufenthalt an der University of British Columbia in Vancouver, Kanada, begann sie 2024 ihre Promotion in der Arbeitsgruppe von Professor Culmsee am Institut für Pharmakologie und Klinische Pharmazie der Philipps-Universität Marburg. Zudem ist sie als Apothekerin in der Apotheke am Ludwigsplatz in Gießen tätig.
Hanna Rosemarie Hofmann studierte von 2019 bis 2024 Pharmazie an der Philipps-Universität Marburg und schließt das Studium im Oktober 2024 mit dem 2. Staatsexamen ab. Im Wahlpflichtfach »Klinische Pharmazie« erstellte sie einen Essay zum Thema »Amyloid-Beta bei Alzheimer-Erkrankung und Impfstoffe gegen Alzheimer«.
Johanna Lücke studierte von 2019 bis 2024 Pharmazie an der Philipps-Universität Marburg und war von 2023 bis 2024 auch als studentische Hilfskraft tätig. Im Oktober 2024 schließt sie das Studium mit dem 2. Staatsexamen ab. Im Wahlpflichtfach »Klinische Pharmazie« erstellte sie einen Essay zum Thema »Neue Behandlungsansätze für Tauopathien bei Morbus Alzheimer«.


