 |  | Theo Dingermann |
 |
15.10.2023 08:00 Uhr |

Arzneimittel für neuartige Therapien (ATMP) werden manchmal sogar als »lebende Arzneimittel« bezeichnet. Können sie schwerste Krankheiten heilen? / Foto: Adobe Stock/natali_mis
Professor Dr. Peter Bader leitet an der Klinik für Kinder- und Jugendmedizin die Abteilung »Stammzelltransplantation, Immunologie und Intensivmedizin«. Täglich erlebt er das Leid und die Verzweiflung seiner kleinen Patienten, die an Leukämie erkrankt sind. Die für diese Patienten oft heilende Stammzelltherapie habe er von der Pike auf gelernt, sagt er in einem Gespräch mit Professor Dr. Manfred Schubert-Zsilavecz anlässlich einer Veranstaltung des House of Pharma and Healthcare (1). Trotz der großen Erfolge der Stammzelltherapie bei Kindern, die an Leukämie erkrankt sind, komme es immer wieder zu Rückschlägen. Bei immunologisch suboptimal passenden Transplantaten ist die Graft-versus-Host-(GvH-)Erkrankung eine der gefürchtetsten Komplikationen. Hier richtet sich das Immunsystem der kleinen Patienten gegen das Implantat und stößt es schließlich ab.
Aus dieser Not heraus habe er immer schon nach optimalen Behandlungsmethoden gesucht, sagt Bader. Und er hatte eine Idee. Bader wusste, dass es eine bestimmte Art von Zellen gibt, die die GvH-Reaktionen abmildern können: sogenannte mesenchymale Stromazellen (MSC). Es gab allerdings einen Haken. Diese Zellen waren therapeutisch kaum brauchbar, da sie je nach Spender mal mehr und mal weniger zuverlässig funktionierten. Es fehlte eine Standardisierung, die reproduzierbare klinische Resultate garantierte.
»Da kam uns die Idee, die Stromazellen von vielen Spendern zu mischen und zusammen zu expandieren«, sagt der Pädiater in dem Gespräch. »Dabei haben sich diese Zellen gegenseitig stimuliert und eine stärkere Potenz entwickelt.«
Das Produkt, das die Frankfurter Wissenschaftler entwickelten, gehört einer relativ neuen Klasse von Arzneimitteln an, die als »Advanced Therapeutic Medicinal Products« (ATMP) bezeichnet werden. Diese Arzneimittel sind nicht mehr durch ein einzelnes Molekül definiert, sondern stellen komplexe Systeme dar, beispielweise lebende Zellen, Vektorsysteme für die Gentherapie oder einen Gewebeverband, bei dem die Zellen in ihre eigene komplexe extrazelluläre Matrix eingebettet sind.
Der Begriff Advanced Therapy Medicinal Products (oder auf Deutsch: Arzneimittel für neuartige Therapien) stammt aus der Zulassungssystematik. Es ist nicht das erste Mal, dass die Zulassungsbehörden für neuartige Produktgruppen ein spezifisches Regelwerk erarbeiten mussten. Anfang dieses Jahrhunderts waren es die Biosimilars, auf die die Zulassungsbehörden reagieren mussten.
Mit den neuartigen Therapien, bei denen das molekulare Wirkprinzip meist durch ein zelluläres und damit hochkomplexes Prinzip abgelöst wird, stellten sich die Regulatoren ein weiteres Mal einer dringenden Herausforderung. Denn aufgrund der Neuheit, Komplexität und technischen Besonderheiten der ATMP mussten eigens auf sie zugeschnittene, harmonisierte Vorschriften erarbeitet werden, um den freien Verkehr dieser Arzneimittel innerhalb der Europäischen Union zu gewährleisten. Zur Arzneistoffklasse der ATMP zählen (2):
Zum Teil sind die Definitionen für die verschiedenen ATMP-Klassen überlappend. Aus diesem Grund hat man sich auf folgende Annahmen verständigt:
Zur wissenschaftlichen Bewertung von Qualität, Wirksamkeit und Unbedenklichkeit einschließlich der Umweltverträglichkeit der neuen Produktgruppe wurde bei der EMA im Jahr 2009 der »Ausschuss für neuartige Therapien« (Committee for Advanced Therapies, CAT) gegründet.
Deutschland wird im CAT durch die Experten des Paul-Ehrlich-Instituts vertreten.
ATMP ergänzen beeindruckend die »konventionellen« Therapieansätze. Mithilfe klassischer Medikamente kann man – mit wenigen Ausnahmen, beispielsweise Antibiotika oder Virustatika – Krankheiten zwar teils hervorragend managen, aber nicht heilen. Dagegen eröffnen ATMP vielfach nicht nur Therapiemöglichkeiten für bisher nicht oder nur unzureichend behandelbare Krankheiten, sondern bieten mit ihrer besonderen Wirkweise auch neue und oft auf Heilung abzielende Optionen für Patienten, die »konventionelle« Therapien ausgeschöpft haben.
Oft leiden die Patienten, die von ATMP profitieren, an seltenen Erkrankungen. Daher werden in die klinischen Studien meist nur wenige Patienten eingeschlossen, sodass für die Zulassung vielfach nur begrenzte Daten zur Verfügung stehen und ein zuverlässiger Nachweis der Wirksamkeit nur schwer erbracht werden kann. Eine weitere Hürde: Da der Einsatz von ATMP oft mit tiefen Eingriffen in den Körper verbunden ist und langfristig auftretende Nebenwirkungen nicht von vornherein ausgeschlossen werden können, werden auch in Phase-I-Studien nur Patienten und keine gesunden Probanden aufgenommen.
Da der medizinische Bedarf in vielen Anwendungsgebieten sehr hoch ist, werden die Produkte mitunter nur bedingt (conditional) oder unter besonderen Umständen (exceptional circumstances) zugelassen.

Viele ATMP werden bei mehrfach vorbehandelten Krebspatienten eingesetzt. / Foto: Adobe Stock/tonefotografia
Derzeit sind in der EU 14 Gentherapeutika, zwei Zelltherapeutika und zwei biotechnologisch bearbeitete Gewebeprodukte zugelassen (3). Darüber hinaus sind Präparate verfügbar, denen das PEI im Rahmen einer §4b-Genehmigung die Verkehrsfähigkeit zuerkannt hat. In diesem Paragrafen des Arzneimittelgesetzes (AMG) sind Sondervorschriften für Arzneimittel für neuartige Therapien geregelt (4). Danach dürfen ATMP, die im Geltungsbereich dieses Gesetzes als individuelle Zubereitung für einen einzelnen Patienten ärztlich verschrieben, nach spezifischen Qualitätsnormen nicht-routinemäßig hergestellt und in einer spezialisierten Einrichtung der Krankenversorgung unter der fachlichen Verantwortung eines Arztes angewendet werden, nur an andere abgegeben werden, wenn sie durch die zuständige Bundesoberbehörde genehmigt worden sind.
Unter den bisher zugelassenen ATMP bilden Gentherapeutika die Hauptgruppe. Ein Gentherapeutikum ist ein biologisches Arzneimittel, dessen Wirkstoff eine Nukleinsäure (Träger der Erbinformationen) enthält oder daraus besteht. Es wird eingesetzt, um eine Nukleinsäure-Sequenz zu regulieren, zu reparieren, zu ersetzen, hinzuzufügen oder zu entfernen. Die therapeutische, prophylaktische oder diagnostische Wirkung steht in unmittelbarem Zusammenhang mit der rekombinanten Nukleinsäure-Sequenz, die es enthält, oder mit dem Protein, das auf Basis dieser genetischen Information gebildet wird (5).
Derzeit sind 14 Fertigarzneimittel von der Europäischen Kommission zugelassen (Grafik). Für weitere drei Präparate (Glybera®, Zynteglo® und Skysona®) haben die Hersteller die Zulassung zurückgegeben. Das ist ein hoher Prozentsatz und deutet Probleme an, die generell mit ATMP verbunden sind.
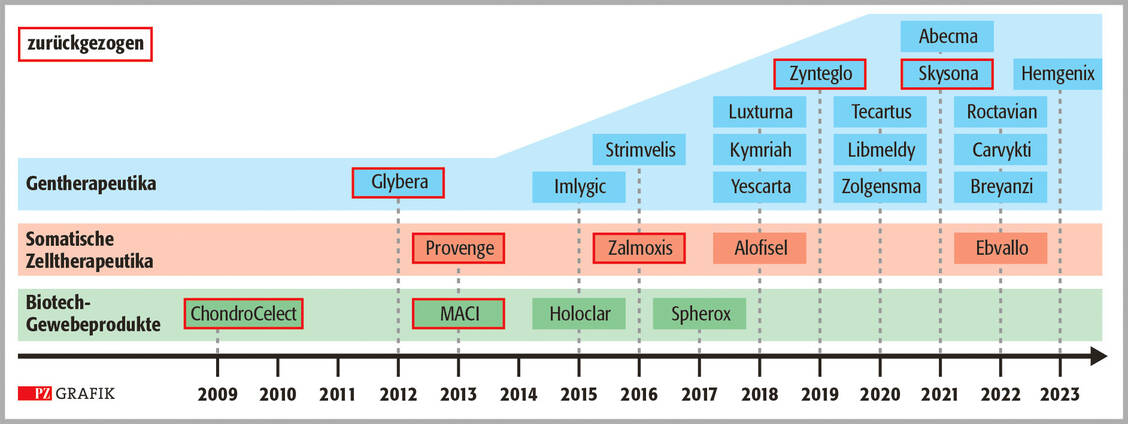
Zeitskala der seit 2009 zentral bei der EMA zugelassenen ATMP (Stand 8/2023); modifiziert nach (6) / Foto: PZ/Stephan Spitzer
Ein lehrreiches Beispiel ist Glybera®, das als erstes Gentherapeutikum bereits 2012 zugelassen wurde (7). Es wurde zur Behandlung von Erwachsenen mit Lipoprotein-Lipase-Defizienz (LPLD) entwickelt, die trotz fettarmer Diät an schweren oder multiplen Entzündungsschüben der Bauchspeicheldrüse leiden. Ursache ist ein Defekt im Gen für das Enzym Lipoprotein-Lipase, das für den Fettabbau verantwortlich ist. Der Wirkstoff von Glybera, Alipogentiparvovec, enthält eine natürlich vorkommende, hochaktive Variante des Gens für die Lipoprotein-Lipase. Dieses Gen wird mithilfe eines viralen Vektors in das Muskelgewebe der Patienten eingeschleust.
Der Weg zur Zulassung von Alipogentiparvovec war lang und »kurvenreich« (8). Es begann mit der Einreichung eines Erstantrags im Dezember 2009, der vom Ausschuss für Humanarzneimittel (CHMP) und vom Ausschuss für neuartige Therapien (CAT) im Juni 2011 abgelehnt wurde. Bei der erneuten Prüfung im selben Jahr blieb einer der beiden Ausschüsse bei seiner ablehnenden Haltung. Als die Europäische Kommission schließlich im Januar 2012 eine erneute Prüfung des Antrags auf Zulassung von Glybera für einen eingeschränkteren Verwendungszweck, nämlich für LPLD-Patienten mit schweren und wiederholten Anfällen von Pankreatitis trotz Einschränkung der Fettaufnahme in der Nahrung, beantragte, gaben sowohl der CHMP als auch der CAT ihre Zustimmung.
Allerdings war die Ernüchterung groß, als erst nach drei langen Jahren die erste zahlende Patientin für das Präparat gefunden wurde. Die Situation verbesserte sich nicht entscheidend, sodass der Hersteller uniQure Glybera nur fünf Jahre später wieder vom Markt nahm.
Hinzu kam, dass die Wirksamkeit von Glybera nie wirklich beeindruckte. Die Blutfettwerte ließen sich nur zeitweilig senken und die Patienten mussten weiterhin strenge Diät halten. Die Zahl der gefährlichen Pankreatitis-Anfälle nahm jedoch spürbar ab und die Betroffenen gewannen ein wichtiges Stück Lebensqualität zurück.
Letztlich scheiterte ein Erfolg jedoch an den hohen Kosten. Die Behandlung an der Berliner Charité schlug mit 900.000 Euro zu Buche. Da die Dosis vom Körpergewicht abhängt, hätten die Kosten bei anderen Patienten leicht die Millionengrenze überschreiten können. Solche Summen wollten die Kassen damals nicht übernehmen.
Dass dieses Beispiel nicht repräsentativ für die klinische Bedeutung der Gentherapeutika ist, lässt sich recht objektiv daran erkennen, dass der Gemeinsame Bundesausschuss (G-BA) mit Stand vom 12. April 2021 nur einem von acht begutachteten Präparaten (Imlygic®) keinen belegbaren Zusatznutzen zuerkennt. Den anderen Präparaten wurden entweder ein »nicht quantifizierbarer Zusatznutzen«, ein »Anhaltspunkt für einen nicht quantifizierbaren Zusatznutzen« oder ein »Anhaltspunkt für einen beträchtlichen Zusatznutzen« zuerkannt (www.covalue.de/gentherapeutika-in-der-frühen-nutzenbewertung).
Eine Erfolgsgeschichte schreiben hingegen die CAR-T-Zell-ATMP (9). CAR-T-Zellen sind gentechnisch veränderte autologe T-Zellen, die einen chimären T-Zellrezeptor exprimieren. Derzeit sind alle CAR-T-Zellpräparate für die Behandlung von Blutkrebs zugelassen, darunter Lymphome, einige Formen von Leukämie und seit Kurzem auch das multiple Myelom.
Das wird aber nicht so bleiben, denn es zeichnen sich CAR-T-Zelltherapeutika ab, die zur Behandlung schwerer Autoimmunerkrankungen, beispielsweise Lupus erythematodes (10), oder schwerer Formen einer Myositis (11) eingesetzt werden.
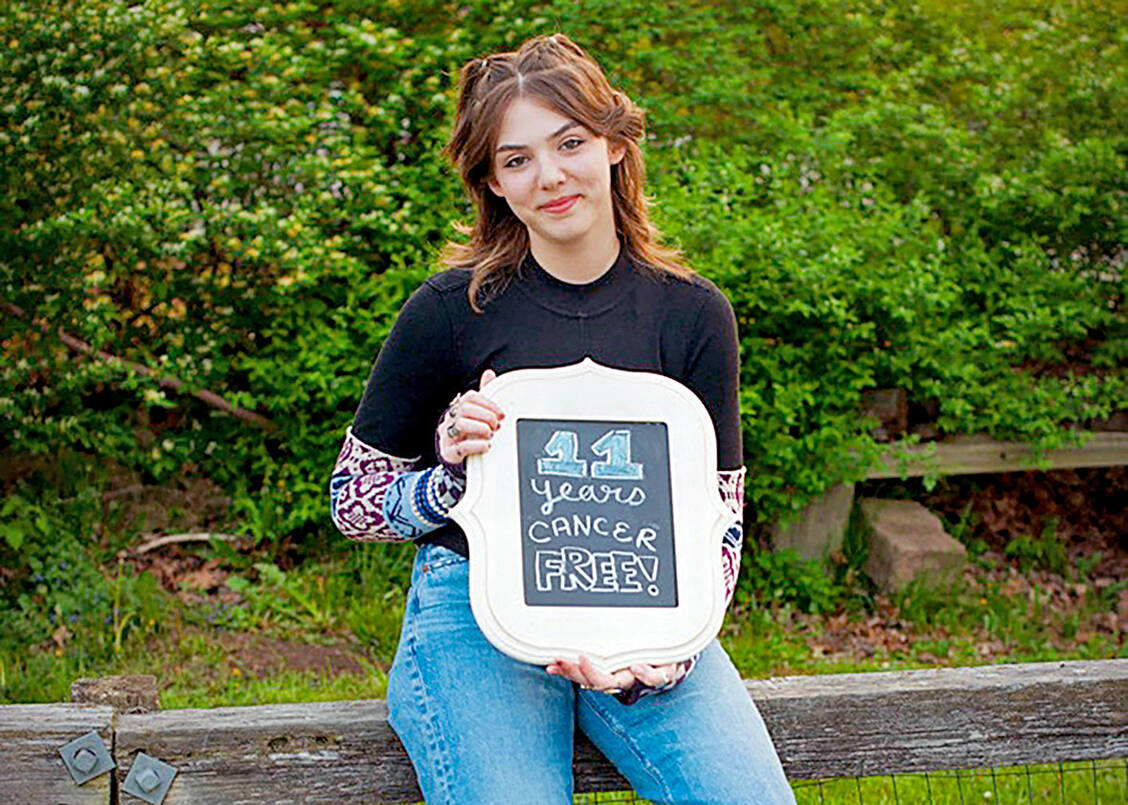
Emily Whitehead litt an akuter lymphoblastischer Leukämie und wurde 2012 als erstes Kind mit einer CAR-T-Zelltherapie behandelt. Sie gilt seitdem als krebsfrei. Ihre Eltern Tom und Kari veröffentlichten jedes Jahr ein Foto ihrer Tochter auf der Website ihrer Stiftung, zum letzten Mal am 10. Mai 2023. Emily ist nun 18 Jahre alt und gilt als geheilt. / Foto: privat
Die chimären T-Zellrezeptoren der meisten zugelassenen CAR-T-Zellpräparate tragen ein Antikörperfragment gegen das CD19-Epitop, das auf der Oberfläche fast aller B-Zellentypen exponiert ist. Diese Präparate erzielen teils bemerkenswerte Therapieerfolge. Beispiele sind Breyanzi®, Kymriah®, Tecartus® und Yescarta®, die alle auf Basis autologer T-Zellen der zu behandelnden Patienten basieren, in die in vitro mithilfe eines lentiviralen Vektors ein chimärer T-Zellrezeptor mit einer CD19-Spezifität eingeschleust wurde (Tabelle1).
Die beiden CAR-T-Zellpräparate Abecma® und Carvykti® enthalten hingegen einen chimären T-Zellrezeptor mit einer CD38-Spezifität. CD38 entspricht dem B-Zell-Reifungsantigen BCMA (B-Cell maturation Antigen). Diese beiden Gentherapeutika sind zur Behandlung eines rezidivierten und refraktären multiplen Myeloms bei erwachsenen Patienten indiziert.
Und dennoch: Trotz der Begeisterung, die diese Therapeutika induziert haben, können sie längst nicht immer die in sie gesetzten Hoffnungen erfüllen. Bei weniger als der Hälfte der behandelten Patienten zeigte die CAR-T-Zelltherapie so weitreichende Erfolge, dass die Patienten nach zwei Jahren immer noch frei von Krebszellen sind.
Noch ernüchternder sind die Erfolgsraten bei einem multiplen Myelom. In einer klinischen Studie zur Wirksamkeit von Abecma® (12) betrug das mittlere progressionsfreie Überleben 13,3 Monate in der Verumgruppe, verglichen mit 4,4 Monaten in der Gruppe mit Standardtherapie (mittlere Nachbeobachtungszeit 18,6 Monaten). Fast drei Viertel der Patienten der CAR-T-Zelltherapie-Gruppe sprachen auf die Behandlung an. Bei 39 Prozent kam es sogar zu einer vollständigen Remission. Dies sind durchaus beachtliche Erfolge, aber keinesfalls Heilversprechen.
| Name, Wirkstoff, Zulassungsinhaber | Art | Indikation |
|---|---|---|
| Abecma®,Idecabtagen vicleucelBristol-Myers Squibb Pharma | CAR-T-Zellen, die das B-Zell-Reifungsantigen (BCMA) erkennen | Drittlinientherapie eines rezidivierten und refraktären multiplen Myeloms |
| Breyanzi®,Lisocabtagen maraleucelBristol-Myers Squibb Pharma | CAR-T-Zellen, die gegen das CD19-Antigen auf B-Zellen gerichtet sind | Zweitlinientherapie eines rezidivierenden oder gegenüber der Erstlinientherapie refraktären DLBCL, eines HGBCL, PMBCL und FL Grad 3B |
| Carvykti®,Ciltacabtagen autoleucelJanssen-Cilag International | CAR-T-Zellen, die das B-Zell-Reifungsantigen (BCMA) erkennen | Viertlinientherapie eines rezidivierten und refraktären multiplen Myeloms |
| Hemgenix®,Etranacogen dezaparvovecCSL Behring GmbH | Gentherapeutikum, das die menschliche Gerinnungsfaktor-IX-Variante R338L (FIX-Padua) unter der Kontrolle eines leberspezifischen Promotors (LP1) codiert | Behandlung einer schweren und mittelschweren Hämophilie B (angeborener Faktor-IX-Mangel) bei Erwachsenen ohne Faktor-IX-Inhibitoren in der Vorgeschichte |
| Imlygic®,Talimogen laherparepvecAmgen Europe | abgeschwächtes Herpes-simplex-Virus Typ 1 (HSV-1), das funktionelle Deletionen von zwei Genen (ICP34.5 und ICP47) trägt und für den humanen Granulozyten-Makrophagen-koloniestimulierenden Faktor codiert | Behandlung von Erwachsenen mit nicht resezierbarem, lokal oder entfernt metastasiertem Melanom (Stadium IIIB, IIIC und IVM1a) ohne Knochen-, Hirn-, Lungen- oder andere viszerale Beteiligung |
| Kymriah®,TisagenlecleucelNovartis Europharm Ltd | CAR-T-Zellen, die gegen das CD19-Antigen auf B-Zellen gerichtet sind | Behandlung von Kindern, Jugendlichen und jungen Erwachsenen (bis 25 Jahre) mit refraktärer oder rezidivierter ALLDrittlinientherapie bei Erwachsenen mit rezidiviertem oder refraktärem DLBCL oder mit rezidiviertem oder refraktärem FL |
| Libmeldy®,Atidarsagen autotemcelOrchard Therapeutics | genetisch veränderte autologe CD34⁺-Zellen, angereichert mit hämatopoetischen Stamm- und Vorläuferzellen (HSPC), die das Gen für humane Arylsulfatase A (ARSA) exprimieren | Behandlung einer biallelischen metachromatischen Leukodystrophie (MLD), die die enzymatische Aktivität von ARSA verringert:• bei Kindern mit im späten Säuglings- oder frühen Kindesalter auftretenden Formen ohne klinische Manifestation,• bei Kindern mit der im frühen Kindesalter auftretenden Form mit frühzeitiger klinischer Manifestation, die noch selbstständig gehen können, vor einer kognitiven Verschlechterung |
| Luxturna®,Voretigen neparvovecNovartis Europharm Ltd | Gentransfer-Vektor, verpackt in nicht replizierendem AAV2, das die cDNA des humanen retinalen Pigmentepithel-spezifischen 65 kDa-Protein (hRPE65) codiert enthält | Behandlung von Erwachsenen und Kindern mit Sehverlust aufgrund einer erblichen Netzhautdystrophie, die auf nachgewiesenen biallelischen RPE65-Mutationen beruht, und die noch ausreichend lebensfähige Netzhautzellen haben |
| Roctavian®,Valoctocogen roxaparvovecBioMarin International Ltd | Gentransfer-Vektor, verpackt in nicht replizierendem AAV5, der die cDNA des Gens für die B-Domänen-deletierte SQ-Form des humanen Gerinnungsfaktors VIII unter der Kontrolle eines leberspezifischen Promoters codiert enthält | Behandlung einer schweren Hämophilie A (kongenitaler Faktor-VIII-Mangel) bei Erwachsenen ohne Faktor-VIII-Inhibitoren in der Anamnese und ohne nachweisbare Antikörper gegen AAV5 |
| Strimvelis®Orchard Therapeutics | autologe CD34⁺-angereicherte Zellfraktion, die CD34⁺-Zellen enthält, die mit einem retroviralen Vektor transduziert wurden, der für die humane Adenosin-Desaminase-(ADA-)cDNA-Sequenz aus humanen hämatopoetischen Stamm-/Progenitorzellen (CD34⁺) codiert | Behandlung eines schweren kombinierten Immundefekts aufgrund von Adenosin-Desaminase-Mangel (ADA-SCID), wenn kein geeigneter HLA-kompatibler Stammzellspender aus der Familie verfügbar ist |
| Tecartus®Brexucabtagen-AutoleucelKite Pharma EU | CAR-T-Zellen, die gegen das CD19-Antigen auf B-Zellen gerichtet sind | Drittlinientherapie bei Erwachsenen mit rezidiviertem oder refraktärem Mantelzell-LymphomBehandlung von Erwachsenen ab 26 Jahren mit rezidivierter oder refraktärer B-Zell-Vorläufer ALL |
| Upstaza®,Eladocagene exuparvovecPTC Therapeutics International Ltd | Gentransfer-Vektor, verpackt in nicht replizierendem AAV2, der die cDNA des humanen Dopa-Decarboxylase-(DDC-)Gens unter der Kontrolle eines CMV-Promoters codiert enthält | Behandlung von Patienten ab 18 Monaten mit einer klinisch, molekularbiologisch und genetisch bestätigten Diagnose eines Aromatische-L-Aminosäure-Decarboxylase-(AADC-)Mangels mit schwerem Phänotyp |
| Yescarta®,Axicabtagen ciloleucelKite Pharma EU | CAR-T-Zellen, die gegen das CD19-Antigen auf B-Zellen gerichtet sind | Zweitlinientherapie von Erwachsenen mit DLBCL und HGBCLDrittlinientherapie von Erwachsenen mit rezidiviertem oder refraktärem (r/r) DLBCL und PMBCLViertlinientherapie von Erwachsenen mit r/r FL |
| Zolgensma®,Onasemnogen-AbeparvovecNovartis Europharm Ltd | Gentransfer-Vektor, verpackt in nicht replizierendem AAV9, der die cDNA des humanen Survival-Motoneuron-(SMN-)Gens unter Kontrolle des CMV-Enhancers/Hühner-β-Aktin-Hybrid-Promotors codiert enthält | Behandlung von Patienten mit 5q-assoziierter spinaler Muskelatrophie (SMA) mit einer biallelischen Mutation im SMN1-Gen und einer klinisch diagnostizierten Typ-1-SMA oder mit bis zu 3 Kopien des SMN2-Gens |
AAVx: Adeno-assoziierter viraler Vektor vom Serotyp x; ALL: akute lymphatische B-Zell-Leukämie; CAR: chimärer Antigenrezeptor; CMV: Cytomegalievirus; DLBCL: diffus großzelliges B-Zell-Lymphom; FL: follikuläres Lymphom; HGBCL: hoch malignes B-Zell-Lymphom; HLA: Human Leukozyten-Antigen; PMBCL: primär mediastinales großzelliges B-Zell-Lymphom
Ein somatisches Zelltherapeutikum besteht aus Zellen oder Geweben, die substanziell bearbeitet wurden, sodass biologische oder strukturelle Merkmale oder physiologische Funktionen verändert wurden. Außerdem kann es aus Zellen oder Geweben bestehen oder diese enthalten, die im Empfänger im Wesentlichen nicht dieselbe Funktion ausüben wie im Spender (fachsprachlich: nicht homologer Gebrauch). Diese Zellen oder Gewebe üben pharmakologische, immunologische oder metabolische Wirkungen aus (13).
Das PEI listet drei somatische Zelltherapeutika auf, die in Deutschland verkehrsfähig sind. Von diesen drei Produkten haben allerdings nur zwei (Alofisel® und Ebvallo®) eine Zulassung durch die Europäische Kommission (Tabelle 2).
| Name, Wirkstoff, Zulassungsinhaber | Art | Indikation |
|---|---|---|
| Alofisel®DarvadstrocelTakeda Pharma | expandierte, humane, allogene, mesenchymale adulte Stammzellen, die aus Fettgewebe (expanded adipose stem cells, eASC) gewonnen wurden | Behandlung von komplexen perianalen Fisteln bei erwachsenen Patienten mit nicht oder gering aktivem luminalen Morbus Crohn |
| Amesanar®Rheacell GmbH & Co. KG(bisher keine EU-Zulassung) | allogene ABCB5-positive mesenchymale Stromazellen | Behandlung von Wunden, zum Beispiel bei chronisch venösem Ulkus, diabetischem Fußulkus oder Ulkus bei peripherer arterieller Verschlusskrankheit |
| Ebvallo®TabelecleucelPierre Fabre Medicament | allogene, für das Epstein-Barr-Virus (EBV) spezifische T-Zell-Immuntherapie, die auf EBV-positive Zellen abzielt und diese unter HLA-(Humanes Leukozyten-Antigen-) Restriktion eliminiert | Zweitlinientherapie bei Erwachsenen und Kindern ab 2 Jahren mit rezidivierter oder refraktärer EBV-positiver Posttransplantations-lymphoproliferativer Erkrankung (PTLD) |
Bei Alofisel® handelt es sich um expandierte, humane, allogene, mesenchymale adulte Stammzellen, die aus Fettgewebe isoliert wurden. Sie sind zur Behandlung von komplexen perianalen Fisteln bei erwachsenen Patienten mit nicht oder gering aktivem luminalen Morbus Crohn indiziert, wenn die Fisteln unzureichend auf mindestens eine konventionelle oder biologische Therapie angesprochen haben. Alofisel® hat auch eine Zulassung zur Behandlung von Wunden, zum Beispiel bei einem chronisch-venösen Ulkus, diabetischen Fußulkus und Ulkus bei peripherer arterieller Verschlusskrankheit.

Menschen mit Morbus Crohn haben oft viele Operationen hinter sich. Bei hartnäckigen perianalen Fisteln kann ihnen ein Zelltherapeutikum helfen. / Foto: Adobe Stock/Martina
Ebvallo® enthält allogene, für das Epstein-Barr-Virus (EBV) spezifische T-Zellen, die auf EBV-positive Zellen abzielen und diese unter HLA-Restriktion eliminieren. Tabelecleucel wird aus T-Zellen hergestellt, die von geeigneten menschlichen Spendern gewonnen werden. Nach der Entnahme werden die T-Zellen ex vivo mit EBV-infizierten B-Zellen derselben Person gemischt und dadurch gegen das Virus sensibilisiert. Wird ein Patient mit EBV-positivem Posttransplantationslymphom mit Tabelecleucel behandelt, greifen die T-Zellen in seinem Körper EBV-infizierte Zellen an und zerstören sie.
Das dritte gelistete Präparat, Amesanar®, hat noch keine Zulassung durch die Europäische Kommission/EMA. Es enthält allogene ABCB5-positive mesenchymale Stammzellen, die aus Hautgewebe isoliert wurden (14). Dies ist eine hochpotente Subpopulation der unter anderem in der Dermis residenten Stammzellen, die an ihrer Oberfläche das Oberflächenprotein ABCB5 (ein Mitglied der ABC-Transporter) exprimieren. ABCB5-positive mesenchymale Stammzellen wirken entzündungshemmend, indem sie in eine Wechselwirkung mit Immunzellen (Makrophagen, T-Zellen, Neutrophile) treten und das Gleichgewicht von einer M1-Makrophagen-dominierten Entzündung hin zu einem M2-Makrophagen-dominierten, heilungsfördernden Gewebeumfeld verschieben. Das formulierte Stammzellpräparat, das als Fertigspritze in einem validierten und temperaturkontrollierten (zwischen 2 und 8 °C) Versandbehältnis geliefert wird, wird auf Wunden aufgetragen, um den Heilungsprozess zu unterstützen.
Ein biotechnologisch bearbeitetes Gewebeprodukt (tissue engineered product, TEP) ist ein biologisches Arzneimittel, das biotechnologisch bearbeitete Zellen oder Gewebe enthält oder daraus besteht. Es dient der Regeneration oder Wiederherstellung oder zum Ersatz menschlichen Gewebes (15).
Das PEI listet acht durch die Europäische Kommission zugelassene oder in Deutschland im Rahmen einer §4b-Genehmigung (AMG) verkehrsfähige Produkte auf (Tabelle 3). Die zugelassenen Produkte sind Holoclar® zur Behandlung einer Limbusstammzellen-Insuffizienz nach Verbrennung/Verätzung des Auges und Spherox® zur Reparatur bestimmter Knorpeldefekte.
| Name, Zulassungsinhaber | Wirkstoff | Art | Indikation |
|---|---|---|---|
| BioSeed®-C autologes 3D-Chondrozytentransplantat, 28,8 Mio. Zellen pro EinheitBioTissue Technologics GmbH | Matrix-gekoppelte humane autologe Chondrozyten | Transplantat humanen Ursprungs | zur Implantation |
| co.don chondrosphere, 10 bis 70 Sphäroide/cm²,Co.don AG | Matrix-assoziierte Zellen in Suspension | Sphäroide aus humanen autologen Matrix-assoziierten Chondrozyten | zur Implantation |
| Holoclar®Holostem Terapie Avanzate (HTA) | lebendes Gewebeäquivalent, 79.000 bis 316.000 Zellen/cm² | ex vivo expandierte autologe menschliche Hornhautepithelzellen, die Stammzellen enthalten | Behandlung von Erwachsenen mit mittelschwerer bis schwerer Limbusstammzellen-Insuffizienz |
| Novocart 3DTetec AG | Matrix-gekoppelte humane autologe Chondrozyten | in vitro expandierte humane autologe Gelenkchondrozyten | zur Implantation |
| Obnitix®Medac Gesellschaft für klinische Spezialpräparate mbH | mesenchymale adulte Stammzellen, extrahiert aus humanem Fettgewebe (expandiert, allogen) | humane allogene mesenchymale Stromazellen, expandiert | Suspension zur intravenösen Infusion |
| Spherox®Co.don AG | 10 bis 70 Sphäroide/cm² | Sphäroide aus humanen autologen Matrix-assoziierten Chondrozyten zur Implantation | Reparatur symptomatischer Gelenkknorpeldefekte der Femurkondyle und der Patella des Knies (ICRS-Grad III oder IV) mit Defektgrößen von bis zu 10 cm² bei Erwachsenen und Jugendlichen mit geschlossener Epiphysenfuge in dem betroffenen Gelenk |
ICRS: International-Cartilage-Regeneration & Joint Preservation Society
An dieser Stelle kommen wir zurück an den Anfang des Artikels. Denn das von Bader und Kollegen entwickelte Stammzellpräparat fällt in diese Gruppe der biotechnologisch bearbeiteten Gewebeprodukte. Zwischenzeitlich konnten die Forschenden das Verfahren bis zur Produktreife entwickeln. Das Medikament, das von der Medac Gesellschaft für klinische Spezialpräparate mbH vertrieben wird, heißt Obnitix® (16). Angewendet wird es zusätzlich zur konventionellen immunsuppressiven Therapie bei steroidrefraktärer akuter Graft-versus-Host-Disease bei Patienten nach einer allogenen Stammzelltransplantation (17). Allerdings befindet sich das Medikament noch immer im europäischen Zulassungsverfahren. Ungeachtet dessen wurde Obnitix® mit dem Innovationspreis der Pharmazeutischen Zeitung 2020 ausgezeichnet (18).
Im August 2021 startete eine große klinische Studie, um die Wirksamkeit und Verträglichkeit von Obnitix® zu evaluieren (IDUNN; NCT04629833). In die randomisierte zweiarmige Phase-III-Studie werden insgesamt circa 210 erwachsene und jugendliche Patienten eingeschlossen und entweder mit Obnitix® oder einer vordefinierten Vergleichstherapie behandelt (BAT: best available therapy). Der primäre Studienendpunkt ist das Gesamtansprechen; zu den sekundären Endpunkten gehören unter anderem das Gesamtüberleben und die Freiheit von Therapieversagen. Erste Ergebnisse der Studie werden für 2025 erwartet.
Tumorimpfstoffe sind therapeutische Impfstoffe, die zur Behandlung von Krebserkrankungen dienen und eine Immunantwort gegen die Krebszellen vermitteln sollen. Damit unterscheiden sie sich von prophylaktischen Impfstoffen zum Schutz vor Infektionskrankheiten wie Masern oder Grippe (19).
Bisher gibt es nur ein Produkt, das indiese ATMP-Kategorie fällt. Dabei handelt es sich um »Zytokin-aktivierte Killerzellen (CIK-Zellen), frisch oder kryokonserviert, allogen«. Dies sind CD3⁺-CD56-T-Zellen, die zu ≤ 1 × 10⁸ Zellen/kg Körpergewicht in ≤ 100 ml Infusionsdispersion appliziert werden. Das Präparat ist vom PEI in Deutschland im Rahmen einer §4b-Genehmigung (AMG) zugelassen (20). Der Zulassungsinhaber ist der DRK-Blutspendedienst Baden-Württemberg.
Das Medikament wird bei Patienten eingesetzt, bei denen nach erfolgter Stammzelltransplantation ein Rezidiv der Leukämie festgestellt wurde (21).
Die bisherige Erfahrung zeigt, dass ein fairer und gleicher Zugang zur medizinischen Versorgung in Europa, die in der »Arzneimittelstrategie für Europa« der Kommission gefordert wird (22), gerade für ATMP nicht gegeben ist. Der in allen EU-Mitgliedstaaten gültigen einheitlichen Zulassung für das Inverkehrbringen steht eine länderspezifische Vielfalt an Kosten-Nutzen-Modellen, Erstattungsmodellen und gesetzlichen Vorgaben gegenüber. Die teils wenig überzeugenden Wirksamkeitsnachweise und eine begrenzte Nachbeobachtungszeit nach einmaliger Gabe in Verbindung mit hohen Kosten und das Fehlen innovativer Preisbildungsmodelle werden als Gründe genannt, weshalb ATMP nicht oder nur verzögert erstattet werden.

Die Europäische Kommission fordert einen fairen und gleichen Zugang zur medizinischen Versorgung und zu Medikamenten in Europa. Das ist bislang keineswegs selbstverständlich. / Foto: Getty Images/kate_sept2004
Tatsächlich scheitert die Verfügbarkeit bestimmter ATMP meist an der Einigung des pharmazeutischen Unternehmens mit den Krankenkassen. Dann geben die Unternehmen die Zulassung für ihre Produkte zurück und verlassen die nationalen Märkte oder den EU-Markt.
Beispiele sind die beiden Gentherapeutika Skysona® und Zytelgo® desUS-amerikanischen Unternehmens Bluebird (23). Skysona® ist das erste Gentherapeutikum zur Behandlung der zerebralen Adrenoleukodystrophie, einer schweren vererbten neurologischen Erkrankung, und Zynteglo® war zur Behandlung einer schweren Beta-Thalassämie zugelassen.
Zu hoffen bleibt, dass der Zugang zu ATMP mit der neuen EU-HTA-Verordnung (HTA: Health Technology Assessment) verbessert wird, die im Jahr 2025 in Kraft treten wird (24). Mit dieser Verordnung wird die Nutzenbewertung von neuen Therapien erstmals auf europäischer Ebene geregelt. Diese erfolgt dann parallel zur europäischen Zulassung. Ziele sind der schnellere Zugang zu neuen Therapien, die Verringerung von Doppelarbeit und die Harmonisierung der klinischen Bewertung.
Theo Dingermann studierte Pharmazie in Erlangen. Nach Promotion und Habilitation war er bis 2013 Geschäftsführender Direktor des Instituts für Pharmazeutische Biologie an der Goethe-Universität Frankfurt am Main. Jetzt ist er Seniorprofessor der Universität. Die Apotheker kennen ihn als Referenten und Autor von wissenschaftlichen Fach- und Lehrbüchern. Der PZ ist er seit April 2010 als externes Mitglied der Chefredaktion, seit Frühjahr 2019 als einer von drei Chefredakteuren und aktuell als Senior Editor verbunden.


